細胞死亡
細胞死亡是生命不可逆停止及生命結束,正常的組織中經常發生細胞死亡,是維持組織機能和形態所必須的。細胞死亡有兩種方式(如圖1所示):一種是細胞凋亡,又稱程序性細胞死亡或細胞自殺,這是細胞死亡的一種生理形式,是細胞在基因控制下主動死亡的過程;另一種是壞死,為一種不受控制的細胞死亡形式,往往在缺血和其他損傷的情況下發生的。
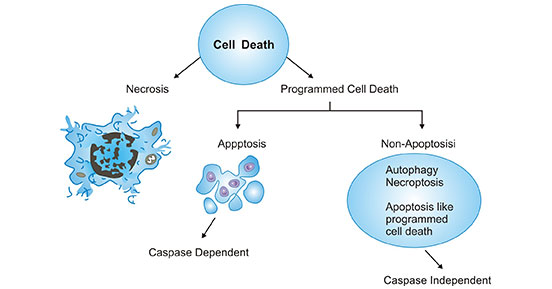
圖1. 細胞死亡的兩種方式
*圖片來源于ResearchGate 的出版物 [1]
近年來,隨著細胞死亡分子機制研究的深入,研究人員發現一些細胞因子可以影響細胞死亡。細胞因子是由免疫細胞和非免疫細胞合成和分泌的小分子肽類因子。本文主要介紹參與細胞凋亡的細胞因子。
細胞凋亡是細胞的一種基本生物學現象,在多細胞生物體內,這對清除不需要的或異常的細胞起著重要作用。細胞凋亡由多個基因嚴格控制,這些基因在物種間非常保守,如Bcl-2家族、caspase家族、C-myc、P53等。參與細胞凋亡的細胞因子主要包括IL-2、IL-3、IL-4、GMSCF、SCF、Epo、TNFɑ和TGFβ等。
IL-2與細胞凋亡
IL-2主要由活化的T細胞產生,能促進T細胞和B細胞的增殖和分化,增強NK細胞和單核細胞的殺傷活性。IL-2還可以抑制多種細胞的凋亡。1993年,相關研究發現,IL-2可以促進CTLL2細胞中原癌基因Bcl-2的表達,Bcl-2是殺傷性T細胞之一。Bcl-2是公認的抑制細胞凋亡的基因,IL-2通過促進Bcl-2的表達來抑制細胞凋亡。此外,CTLL2細胞是IL-2依賴性T細胞。在培養過程中除去IL-2,凋亡會很快發生,而加入IL-2可以抑制凋亡的發生。
IL-3與細胞凋亡
IL-3又稱多聚落刺激因子,主要由活化的T細胞產生,可刺激多能干細胞的增殖和分化。1993年,Mekori發現IL-3可以抑制IL-3依賴性肥大細胞的凋亡。骨髓源性肥大細胞系BMCMC和生長因子依賴性肥大細胞系MCP5都是IL-3依賴性細胞。去除IL-3可導致細胞凋亡,證實IL-3可防止肥大細胞凋亡。
IL-4與細胞凋亡
IL-4主要由活化的T細胞產生。它對B細胞、T細胞肥大細胞、巨噬細胞和造血細胞有免疫調節作用。1996年,Zamorano發現IL-4可以通過胰島素受體底物(IRS)途徑保護細胞凋亡,研究發現IL-4可以介導IRS的磷酸化,從而增加磷脂酰肌醇-3和激酶的活性,抑制細胞凋亡。
GM-CSF與細胞凋亡
GM-CSF主要由T細胞、B細胞、巨噬細胞和成纖維細胞產生。1996年,WeiE發現多形核白細胞(PMN)單獨培養時會迅速凋亡,與GM-CSF一起培養時凋亡明顯減少。PMN中有一個Src家族的酪氨酸蛋白激酶。Lyn是其成員之一,而GM-CSF可以激活Lyn。
SCF和細胞凋亡
SCF又稱肥大細胞生長因子,主要由干細胞產生。它能促進IL-3依賴性的早期造血前體細胞增殖和分化,促進肥大細胞增殖。早期研究發現,SCF可以抑制肥大細胞的凋亡。肥大細胞可以表達c-kit,是SCF的受體。SCF與肥大細胞表面的c-kit結合后,可激活其酪氨酸激酶,從而抑制細胞凋亡。
Epo和細胞凋亡
Epo主要由腎細胞產生,可刺激造血祖細胞的分化和發育。在紅細胞生成過程中,有兩個階段依賴于Epo,包括紅細胞樣集落形成單位(CFUE)階段和原紅細胞階段。1990年,Koury從感染了致貧血病毒的小鼠中分離出FAV細胞(與紅細胞造血祖細胞同種,Epo依賴性與正常CFUE相似)。當Epo在體外培養時,FAV細胞能存活并分化為網狀細胞。反之,FAV細胞的DNA斷裂會導致細胞凋亡。
TNFɑ和細胞凋亡
TNFɑ主要由單核細胞和巨噬細胞產生。1995年,Martin發現TNFɑ和干擾素有促進細胞凋亡的協同作用。HT-29細胞來源于結腸癌,是分化良好的上皮細胞,細胞表面可表達TNFɑ受體。干擾素能促進TNFɑ受體的表達,促進細胞凋亡。
TGFβ與細胞凋亡
TGFβ可由于多種細胞分泌,在細胞生長分化和免疫功能中起著重要作用。TGFβ不僅能促進部分細胞的凋亡,還能抑制部分細胞的凋亡。1995年,Lomo發現TGFβ可以促進成熟B細胞的凋亡。當新鮮分離的外周血B細胞在體外培養時,其凋亡速度緩慢,且速度穩定。當人TGFβ與B細胞共培養時,凋亡明顯增加,且與劑量成正比。但有研究表明,TGFβ可抑制活化T細胞凋亡。TGFβ可以增強T細胞對Fas介導的細胞凋亡的耐受性,延緩細胞由抗凋亡到對凋亡敏感的轉化,從而抑制細胞凋亡。
參考文獻:
[1] Jan, Rehmat & Chaudhry, Gul-e-Saba. (2019). Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Advanced Pharmaceutical Bulletin. 9. 205-218. 10.15171/apb.2019.024.





