Recombinant Human Paraplegin (SPG7)
-
中文名稱:人SPG7重組蛋白
-
貨號:CSB-CF892174HU
-
規格:
-
來源:in vitro E.coli expression system
-
其他:
產品詳情
-
基因名:SPG7
-
Uniprot No.:
-
別名:SPG7; CAR; CMAR; PGN; Paraplegin; Cell matrix adhesion regulator; Spastic paraplegia 7 protein
-
種屬:Homo sapiens (Human)
-
蛋白長度:full length protein
-
表達區域:1-795
-
氨基酸序列MAVLLLLLRALRRGPGPGPRPLWGPGPAWSPGFPARPGRGRPYMASRPPGDLAEAGGRAL QSLQLRLLTPTFEGINGLLLKQHLVQNPVRLWQLLGGTFYFNTSRLKQKNKEKDKSKGKA PEEDEEERRRRERDDQMYRERLRTLLVIAVVMSLLNALSTSGGSISWNDFVHEMLAKGEV QRVQVVPESDVVEVYLHPGAVVFGRPRLALMYRMQVANIDKFEEKLRAAEDELNIEAKDR IPVSYKRTGFFGNALYSVGMTAVGLAILWYVFRLAGMTGREGGFSAFNQLKMARFTIVDG KMGKGVSFKDVAGMHEAKLEVREFVDYLKSPERFLQLGAKVPKGALLLGPPGCGKTLLAK AVATEAQVPFLAMAGPEFVEVIGGLGAARVRSLFKEARARAPCIVYIDEIDAVGKKRSTT MSGFSNTEEEQTLNQLLVEMDGMGTTDHVIVLASTNRADILDGALMRPGRLDRHVFIDLP TLQERREIFEQHLKSLKLTQSSTFYSQRLAELTPGFSGADIANICNEAALHAAREGHTSV HTLNFEYAVERVLAGTAKKSKILSKEEQKVVAFHESGHALVGWMLEHTEAVMKVSITPRT NAALGFAQMLPRDQHLFTKEQLFERMCMALGGRASEALSFNEVTSGAQDDLRKVTRIAYS MVKQFGMAPGIGPISFPEAQEGLMGIGRRPFSQGLQQMMDHEARLLVAKAYRHTEKVLQD NLDKLQALANALLEKEVINYEDIEALIGPPPHGPKKMIAPQRWIDAQREKQDLGEEETEE TQQPPLGGEEPTWPK
Note: The complete sequence may include tag sequence, target protein sequence, linker sequence and extra sequence that is translated with the protein sequence for the purpose(s) of secretion, stability, solubility, etc.
If the exact amino acid sequence of this recombinant protein is critical to your application, please explicitly request the full and complete sequence of this protein before ordering. -
蛋白標簽:N-terminal 10xHis-tagged
-
產品提供形式:Liquid or Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
緩沖液:Lyophilized from Tris/PBS-based buffer, 6% Trehalose, pH 8.0
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet & COA:Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:ATP-dependent zinc metalloprotease. Plays a role in the formation and regulation of the mitochondrial permeability transition pore (mPTP) and its proteolytic activity is dispensable for this function.
-
基因功能參考文獻:
- Compound heterozygous variants in SPG7 identified in 22 French Canadian patients with spastic ataxia. PMID: 26626314
- CACNA1A and SPG7 are major ataxia genes. PMID: 28444220
- The results of this study showed that the most frequently detected variant in this cohort was the SPG7 p.Leu78. PMID: 27084228
- A Norwegian founder mutation p.H701P is a major cause of SPG7 in Norway. PMID: 26756429
- a novel homozygous frameshift deletion in the SPG7 gene was identifies as the genetic cause of hereditary spastic paraplegia in a Greek family. PMID: 26260707
- this case shows that the spectrum of pathologies in SPG7 can include neuron loss of the dentate nucleus and the inferior olivary nucleus as well as neuritic pathology. PMID: 26506339
- Data indicates that SPG7 is essential for the mitochondrial permeability transition pore (PTP) complex formation, interacts with CypD and VDAC and determines C terminus of SPG7 and CsA-binding region of CypD as necessary for PTP formation. PMID: 26387735
- In unexplained ataxia, there was a significant number of patients with SPG7 mutations. PMID: 25681447
- The SPG7 Q866 variant is efficiently processed independent of phosphorylation of AFG3L2 at Y179, which inhibits processing of SPG7. PMID: 24767997
- Using an unbiased exome sequencing approach we identified pathogenic compound heterozygous SPG7 mutations in patients with PEO and multiple mitochondrial DNA deletions in skeletal muscle PMID: 24727571
- A Japanese patient is reported with an SPG7 mutation for a slowly progressive form of autosomal recessive cerebellar ataxia and spastic paraplegia. PMID: 23857099
- This study showed that the p.Ala510Val mutation is prevalent amongst severe hereditary spastic paraparesis patients of UK. PMID: 23269439
- Data suggest a pathogenic role for this SPG7 p.A510V variant. PMID: 22571692
- SPG7 mutations correlate with spastic paraplegia phenotypes. PMID: 22964162
- SPG7 mutations are a frequent cause of middle-aged onset of spastic gait when strict inclusion criteria are applied and should, therefore, be tested in autosomal recessive or sporadic hereditary spastic paraplegia. PMID: 23065789
- Studies indicate that both mouse and human SPG7 ESTs containing alternative first exons. PMID: 22563492
- The novel mutation is the first splice site mutation found in the SPG7 gene. It removes part of the AAA domain of paraplegin protein, probably leading to a loss-of-function of the paraplegin-AFG3L2 complex in the mitochondrial inner membrane. PMID: 20108356
- structural analysis of the ATPase domain of the human AAA+ protein paraplegin/SPG7 PMID: 19841671
- Adenoassociated virus-mediated intramuscular delivery of paraplegin halted the progression of neuropathological changes and rescued mitochondrial morphology in the peripheral nerves of paraplegin-deficient mice. PMID: 16357941
- Cerebellar signs or cerebellar atrophy on brain imaging were the most frequent additional features in patients with SPG7 hereditary spastic paraplegia. PMID: 16534102
- The new SPG7 gene mutation leads to a novel complicated autosomal recessive hereditary spastic paraparesis phenotype that widens the spectrum of different brain systems that are optionally affected in hereditary spastic paraplegia. PMID: 17646629
- identification of six novel point mutations and one large intragenic deletion in hereditary spastic paraplegia PMID: 18200586
- This study identified a novel paraplegin mutation, c.1047insC, in a non-consanguineous Norwegian family with ARHSP. PMID: 18563470
- Results suggest that paraplegin mutations are a frequent cause of sporadic spastic paraparesis. PMID: 18799786
- An intersubunit signaling network coordinates ATP hydrolysis by m-AAA protease paraplegin. PMID: 19748354
顯示更多
收起更多
-
相關疾病:Spastic paraplegia 7, autosomal recessive (SPG7)
-
亞細胞定位:Mitochondrion inner membrane; Multi-pass membrane protein.
-
蛋白家族:AAA ATPase family; Peptidase M41 family
-
組織特異性:Ubiquitous.
-
數據庫鏈接:
Most popular with customers
-
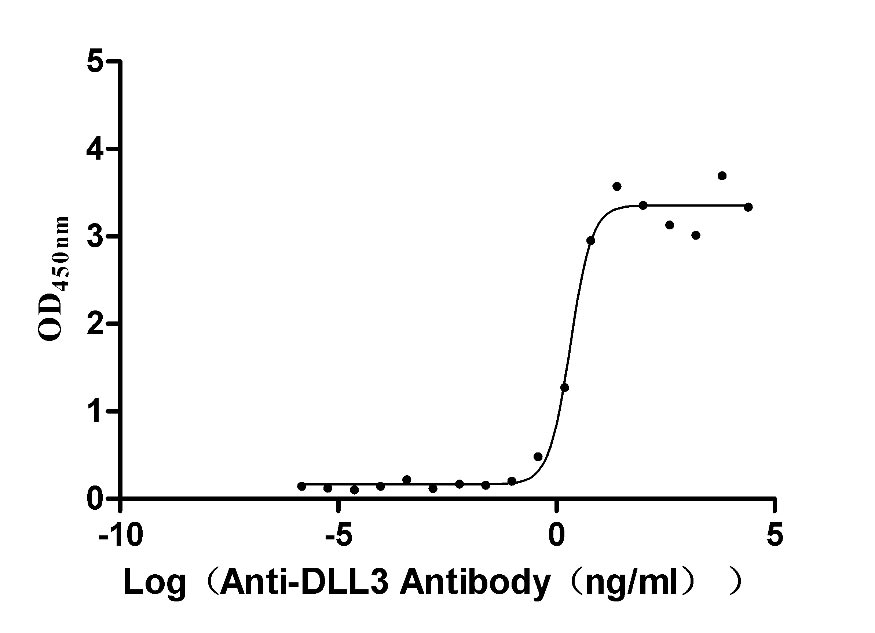
Recombinant Macaca fascicularis Delta-like protein 3 (DLL3), partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
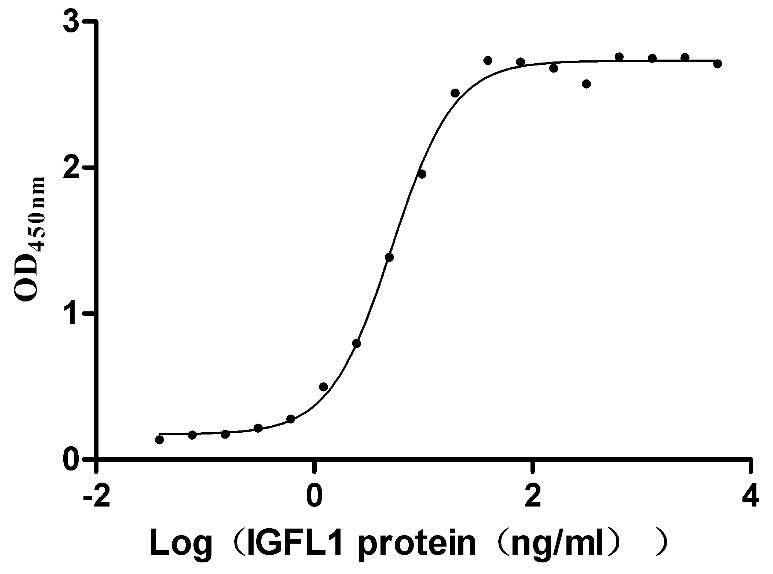
Recombinant Human IGF-like family receptor 1 (IGFLR1), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
-AC1.jpg)
Recombinant Human Claudin-6 (CLDN6)-VLPs (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
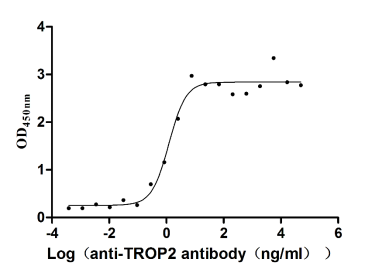
Recombinant Human Tumor-associated calcium signal transducer 2 (TACSTD2), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
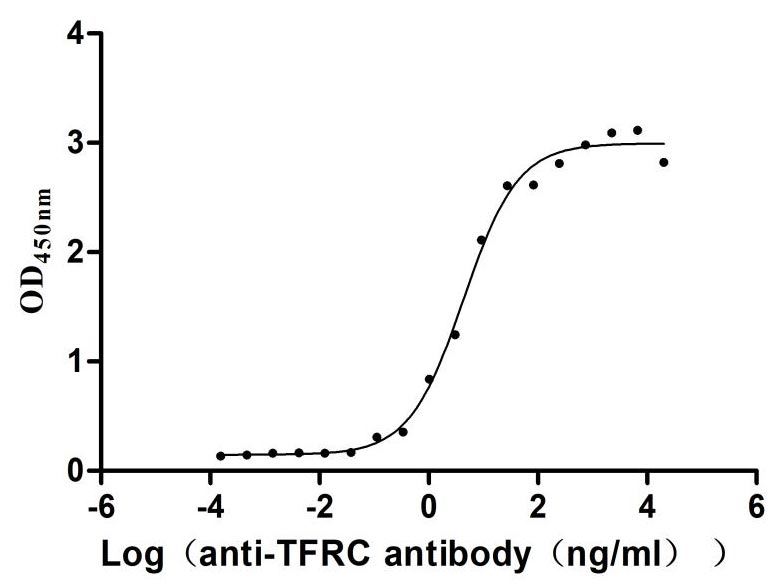
Recombinant Human Transferrin receptor protein 1 (TFRC), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Human Cadherin-1(CDH1),partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Macaca fascicularis Cadherin 6(CDH6),partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Express system: Mammalian cell
Species: Macaca mulatta (Rhesus macaque)









