Recombinant Human Ribitol-5-phosphate transferase FKTN (FKTN)
-
中文名稱:人FKTN重組蛋白
-
貨號:CSB-CF008709HU
-
規格:¥9720
-
圖片:
-
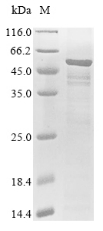 (Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
(Tris-Glycine gel) Discontinuous SDS-PAGE (reduced) with 5% enrichment gel and 15% separation gel.
-
-
其他:
產品詳情
-
純度:Greater than 85% as determined by SDS-PAGE.
-
基因名:FKTN
-
Uniprot No.:
-
別名:FKTN; FCMD; Fukutin; Fukuyama-type congenital muscular dystrophy protein; Ribitol-5-phosphate transferase
-
種屬:Homo sapiens (Human)
-
蛋白長度:Full Length
-
來源:in vitro E.coli expression system
-
分子量:56.5 kDa
-
表達區域:1-461aa
-
氨基酸序列MSRINKNVVLALLTLTSSAFLLFQLYYYKHYLSTKNGAGLSKSKGSRIGFDSTQWRAVKKFIMLTSNQNVPVFLIDPLILELINKNFEQVKNTSHGSTSQCKFFCVPRDFTAFALQYHLWKNEEGWFRIAENMGFQCLKIESKDPRLDGIDSLSGTEIPLHYICKLATHAIHLVVFHERSGNYLWHGHLRLKEHIDRKFVPFRKLQFGRYPGAFDRPELQQVTVDGLEVLIPKDPMHFVEEVPHSRFIECRYKEARAFFQQYLDDNTVEAVAFRKSAKELLQLAAKTLNKLGVPFWLSSGTCLGWYRQCNIIPYSKDVDLGIFIQDYKSDIILAFQDAGLPLKHKFGKVEDSLELSFQGKDDVKLDVFFFYEETDHMWNGGTQAKTGKKFKYLFPKFTLCWTEFVDMKVHVPCETLEYIEANYGKTWKIPVKTWDWKRSPPNVQPNGIWPISEWDEVIQLY
Note: The complete sequence may include tag sequence, target protein sequence, linker sequence and extra sequence that is translated with the protein sequence for the purpose(s) of secretion, stability, solubility, etc.
If the exact amino acid sequence of this recombinant protein is critical to your application, please explicitly request the full and complete sequence of this protein before ordering. -
蛋白標簽:N-terminal 10xHis-tagged
-
產品提供形式:Liquid or Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
緩沖液:If the delivery form is liquid, the default storage buffer is Tris/PBS-based buffer, 5%-50% glycerol.If the delivery form is lyophilized powder, the buffer before lyophilization is Tris/PBS-based buffer, 6% Trehalose.
-
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20°C/-80°C. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet & COA:Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:Catalyzes the transfer of CDP-ribitol to the distal N-acetylgalactosamine of the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1). This constitutes the first step in the formation of the ribitol 5-phosphate tandem repeat which links the phosphorylated O-mannosyl trisaccharide to the ligand binding moiety composed of repeats of 3-xylosyl-alpha-1,3-glucuronic acid-beta-1. Required for normal location of POMGNT1 in Golgi membranes, and for normal POMGNT1 activity. May interact with and reinforce a large complex encompassing the outside and inside of muscle membranes. Could be involved in brain development (Probable).
-
基因功能參考文獻:
- The results suggest that fukutin and FKRP not only participate in the synthesis of O-mannosyl glycans added to alpha-dystroglycan in the endoplasmic reticulum and Golgi complex, but that they could also play a role, that remains to be established, in the nucleus of retinal neurons. PMID: 29416295
- ISPD and FKTN are essential for the incorporation of ribitol into alpha-dystroglycan. PMID: 27194101
- the mutated fukutin protein was smaller than the normal protein, reflecting the truncation of fukutin due to a premature stop codon. Immunostaining analysis showed a decrease in the signal for the glycosylated form of alpha-dystroglycan. These findings indicated that this mutation is the second most prevalent loss-of-function mutation in Japanese Fukuyama congenital muscular dystrophy patients. PMID: 28680109
- Fukutin, FKRP, and TMEM5 form a complex while maintaining each of their enzyme activities. Data showed that endogenous fukutin and FKRP enzyme activities coexist with TMEM5 enzyme activity, and suggest the possibility that formation of this enzyme complex may contribute to specific and prompt biosynthesis of glycans that are required for dystroglycan function. PMID: 29477842
- Fukutin and fukutin-related protein are sequentially acting Rbo5P transferases that use cytidine diphosphate ribitol. PMID: 26923585
- Fukutin role in in tumor progression in gastric cancer PMID: 26223471
- Mutation in the fukutin gene is associated with Fukuyama congenital muscular dystrophy and microcephaly. PMID: 24530477
- four new non-Japanese patients with FKTN mutations and congenital muscular dystrophy PMID: 20961758
- FKTN mutations are the most common genetic cause of congenital muscular dystrophies with defective alpha-dystroglycan glycosylation in Korea PMID: 20620061
- In Fukuyama congenital muscular dystrophy (FCMD) cases, expression of fukutin looked decreased. PMID: 12172906
- Fukutin is associated with Walker-Warburg syndrome. PMID: 14627679
- Data suggest that fukutin and fukutin-related protein (FKRP) may be involved at different steps in O-mannosylglycan synthesis of alpha-dystroglycan, and FKRP is most likely involved in the initial step in this synthesis. PMID: 15213246
- Fukutin seems to bind to both the hypoglycosylated and fully glycosylated form of alpha-dystroglycan, and seems bind to the core area rather than the sugar chain of alpha-dystroglycan PMID: 17005282
- Walker-Warburg syndrome carries a homozygous-single nucleotide insertion that produces a frameshift, or 2 mutations, a point mutation that produces an amino acid substitution, & deletion in 3'UTR that affects the polyadenylation signal of fukutin gene. PMID: 18177472
- FCMD mutations are a more common cause of Walker-Warburg syndrome outside of the Middle East. PMID: 18752264
- The homozygous nonsense mutations within the coding region identified in Turkish patients are predicted to cause a total loss of fukutin activity and are likely to produce a more severe phenotype which closely resembles WWS. PMID: 18834683
- The compound heterozygous FKTN mutation was a rare cause of dilated cardiomyopathy. Hyper-CKemia might be indicative of FKTN mutation in dilated cardiomyopathy. PMID: 19015585
- Outside Japan, fukutinopathies are associated with a large spectrum of phenotypes from isolated hyperCKaemia to severe CMD, showing a clear overlap with that of FKRP. PMID: 19179078
- an identical homozygous c.1167insA mutation in the FKTN gene on a common haplotype in four families and identified 2/299 (0.7%) carriers for the c.1167insA mutation among normal American Ashkenazi Jewish adults PMID: 19266496
- Our results provide further evidence for ethnic and allelic heterogeneity and the presence of milder phenotypes in FKTN-dystroglycanopathy despite a substantial degree of alpha-dystroglycan hypoglycosylation in skeletal muscle. PMID: 19342235
- We found fukutin gene mutations in a 4.5-year-old Italian patient, with reduced alpha-dystroglycan expression, dystrophic features on muscle biopsy, hypotonia since birth, mild myopathy, but no brain involvement. PMID: 19396839
顯示更多
收起更多
-
相關疾病:Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A4 (MDDGA4); Muscular dystrophy-dystroglycanopathy congenital without mental retardation B4 (MDDGB4); Muscular dystrophy-dystroglycanopathy limb-girdle C4 (MDDGC4); Cardiomyopathy, dilated 1X (CMD1X)
-
亞細胞定位:Golgi apparatus membrane; Single-pass type II membrane protein. Cytoplasm. Nucleus.
-
蛋白家族:LicD transferase family
-
組織特異性:Expressed in the retina (at protein level). Widely expressed with highest expression in brain, heart, pancreas and skeletal muscle. Expressed at similar levels in control fetal and adult brain. Expressed in migrating neurons, including Cajar-Retzius cells
-
數據庫鏈接:
Most popular with customers
-
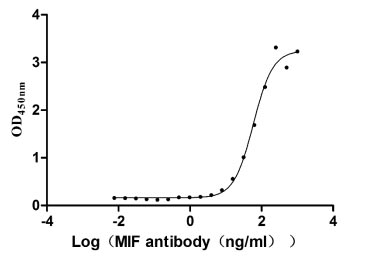
Recombinant Human Macrophage migration inhibitory factor (MIF) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
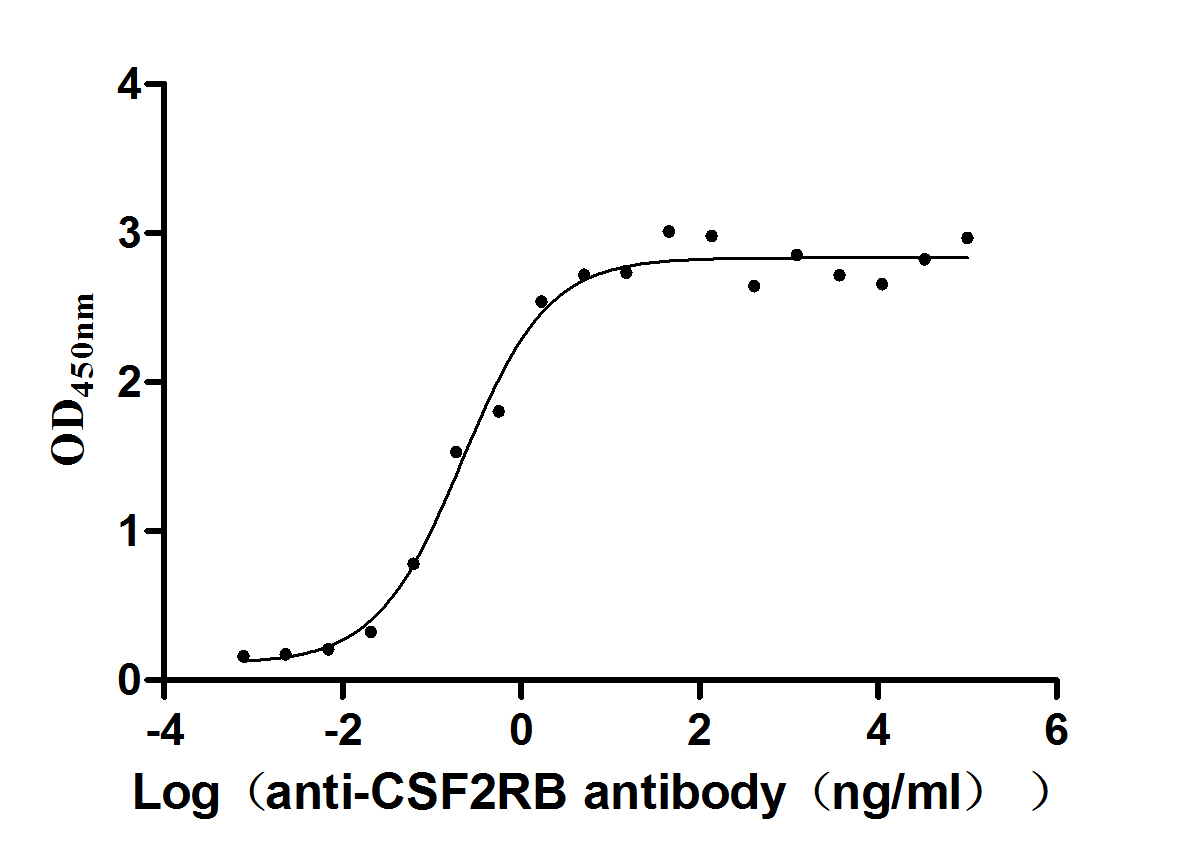
Recombinant Human Cytokine receptor common subunit beta (CSF2RB), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
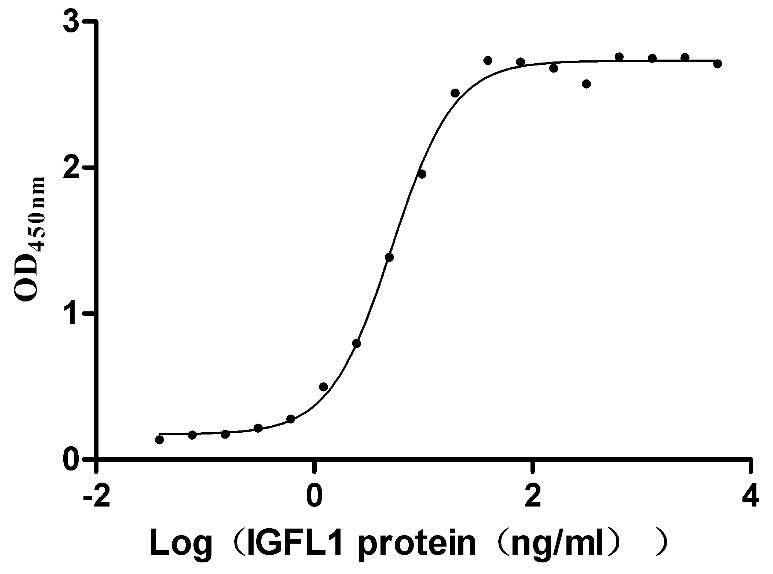
Recombinant Human IGF-like family receptor 1 (IGFLR1), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
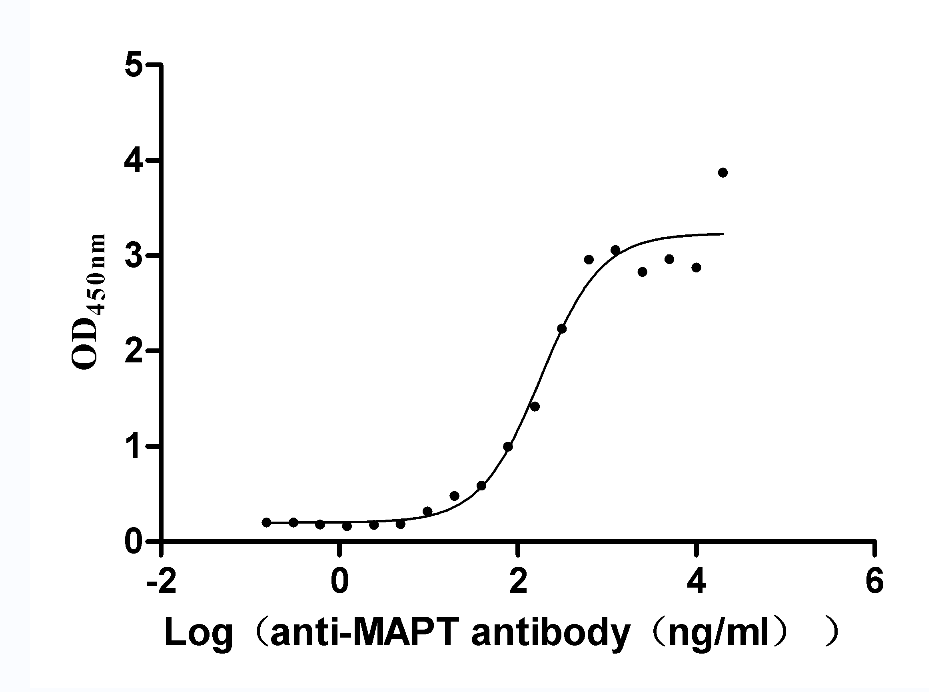
Recombinant Rat Microtubule-associated protein tau (Mapt) (Active)
Express system: Mammalian cell
Species: Rattus norvegicus (Rat)
-
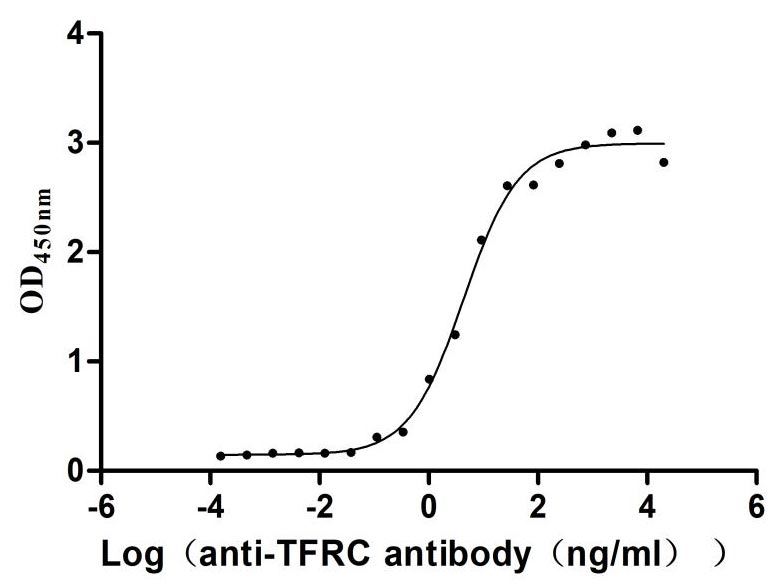
Recombinant Human Transferrin receptor protein 1 (TFRC), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
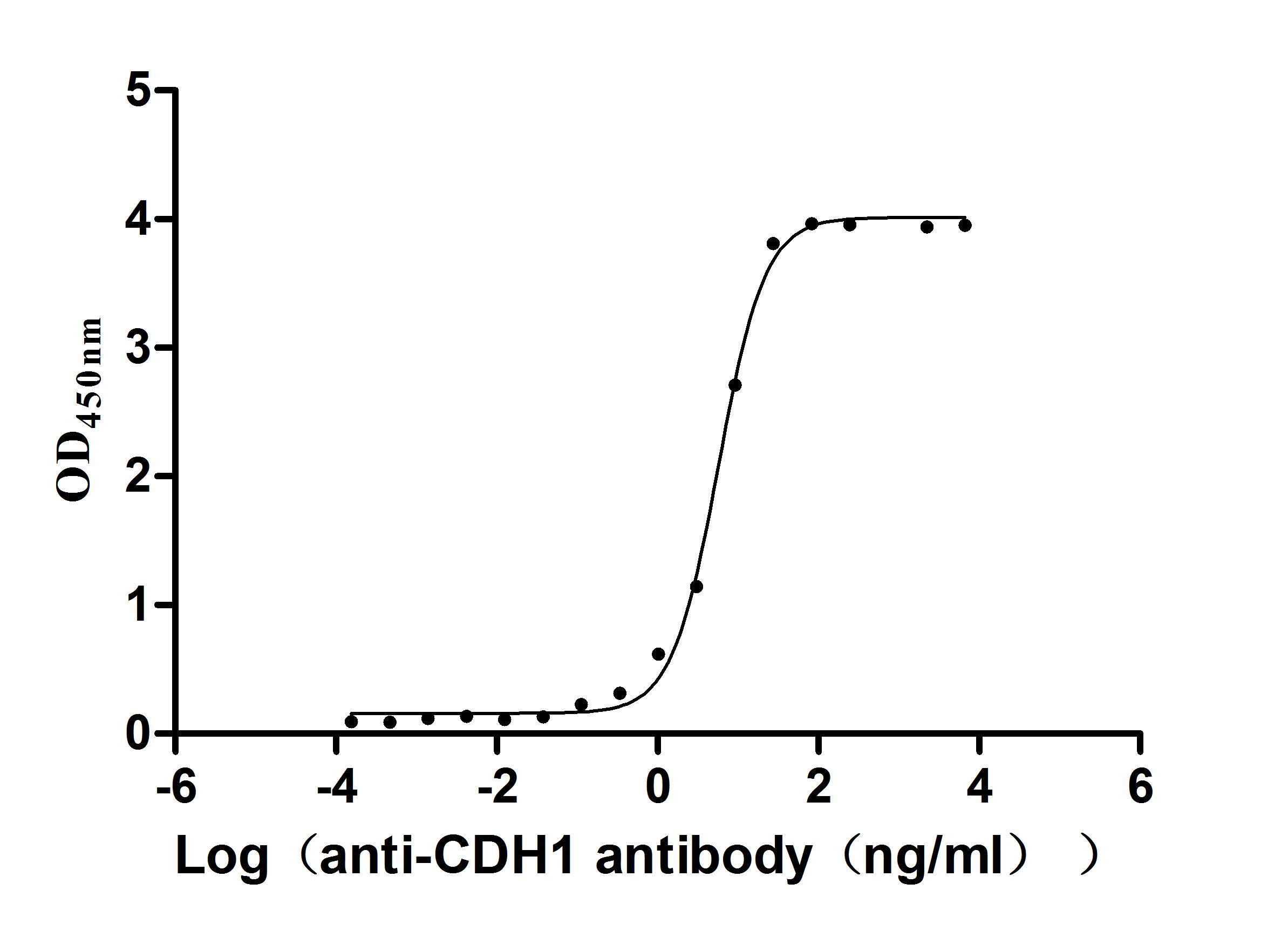
Recombinant Human Cadherin-1(CDH1),partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)









