Recombinant Human Sodium bicarbonate transporter-like protein 11 (SLC4A11), partial
-
中文名稱:人SLC4A11重組蛋白
-
貨號:CSB-EP818756HU-B
-
說明書:
-
規格:
-
來源:E.coli
-
共軛:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
產品詳情
-
純度:>85% (SDS-PAGE)
-
基因名:SLC4A11
-
Uniprot No.:
-
別名:Bicarbonate transporter related protein 1; Bicarbonate transporter-related protein 1; BTR1; CDPD; CHED2; Corneal endothelial dystrophy 2 autosomal recessive; dJ794I6.2; FECD4; MGC126418; MGC126419; NaBC1; S4A11_HUMAN; Slc4a11; Sodium bicarbonate transporter-like protein 11; Sodium borate cotransporter 1; Sodium coupled borate cotransporter 1; Solute carrier family 4 member 11; Solute carrier family 4 sodium bicarbonate transporter like member 11; Solute carrier family 4 sodium borate transporter member 11
-
種屬:Homo sapiens (Human)
-
蛋白長度:Partial
-
蛋白標簽:Tag?type?will?be?determined?during?the?manufacturing?process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
產品提供形式:Lyophilized powder Warning: in_array() expects parameter 2 to be array, null given in /www/web/cusabio_cn/public_html/caches/caches_template/default/content/show_product_protein.php on line 662
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
復溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
儲存條件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保質期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
貨期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事項:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
產品評價

相關產品
靶點詳情
-
功能:Multifunctional transporter with an impact in cell morphology and differentiation. In the presence of borate B(OH)4(-), acts as a voltage-dependent electrogenic Na(+)-coupled B(OH)4(-) cotransporter controlling boron homeostasis. At early stages of stem cell differentiation, participates in synergy with ITGA5-ITGB1 and ITGAV-ITGB3 integrins and BMPR1A to promote cell adhesion and contractility that drives differentiation toward osteogenic commitment while inhibiting adipogenesis. In the absence of B(OH)4(-), acts as a Na(+)-coupled OH(-) or H(+) permeable channel with implications in cellular redox balance. Regulates the oxidative stress response in corneal endothelium by enhancing antioxidant defenses and protecting cells from reactive oxygen species. In response to hypo-osmotic challenge, also acts as water permeable channel at the basolateral cell membrane of corneal endothelial cells and facilitates transendothelial fluid reabsorption in the aqueous humor. In the presence of ammonia, acts as an electrogenic NH3/H(+) cotransporter and may play a role in ammonia transport and reabsorption in renal Henle's loop epithelium.
-
基因功能參考文獻:
- Based on these findings, we infer that high SLC4A11 expression is an independent predictor for poor OS in grade 3/4 serous ovarian cancer. Both DNA amplification and hypomethylation contribute to its upregulation in ovarian cancer PMID: 29091960
- A missense SLC4A11 mutation (Leu843Pro) is responsible for CHED2 in this family; this is the first report of this mutation in a homozygous state. PMID: 27057589
- for the first time, compound heterozygous SLC4A11 mutations impair protein function leading to delayed onset of the disease. PMID: 27609159
- These complex ion transport properties need to be addressed in the context of corneal endothelial disease processes caused by mutations in SLC4A11. PMID: 27581649
- A role of human SLC4A11 in bicarbonate or borate transport. PMID: 27558157
- Slc4a11 is an ideally selective H(+)/OH(-) conductive pathway. PMID: 27681179
- SLC4A11 rs3810560 polymorphism independently affected the sustained viral response rates in chronic hepatitis C patients treated with PEGIFN2b/ribavirin/combination. PMID: 26750805
- Analysis of SLC4A11, ZEB1, LOXHD1, COL8A2 and TCF4 gene sequences in a multi-generational family with late-onset Fuchs corneal dystrophy found no evidence for found polymorophisms causing the disease in this specific pedigree. PMID: 27121161
- study reports a newly identified mutation (c.2024A>C) in the SLC4A11 gene segregating with the diseased haplotype in two consanguineous Pakistani families PMID: 26286922
- we report posterior polymorphous corneal dystrophy resulting from a de novo mutation in ZEB1. Additionally, we present a congenital hereditary endothelial dystrophy case with a thin Descemet membrane with a novel compound heterozygous SLC4A11 mutation. PMID: 26619383
- we review the current knowledge on the role of the SLC4A11 gene, protein, and its mutations in the pathophysiology and clinical presentation of CHED. [review] PMID: 26451371
- We report a novel nonsense mutation of the SLC4A11 gene in the patient with CHED2. In addition, one of heterozygous carriers in this family showed features of late onset Fuchs endothelial corneal dystrophy. PMID: 24502824
- SLC4A11 mutations contribute to 11% (5/45) of sporadic late-onset Late-onset Fuchs endothelial corneal dystrophy (FECD) in the cohort studied. PMID: 25007886
- SLC4A11 is a novel NH3/H+ co-transporter. PMID: 26018076
- We found that cells containing mutant SLC4A11 are more vulnerable to oxidative and mitochondrial damage, less able to overcome oxidative stress through the expression of sufficient levels of antioxidant genes, and are more prone to apoptotic death. PMID: 25811729
- In contrast to the Slc4a11(-/-) mouse, no abnormalities in daily renal ion excretion or polyuria were observed in the Harboyan syndrome patient. PMID: 25500497
- Potential therapeutic agents to improve the functional impairment of specific SLC4A11 mutant transporters. PMID: 25394471
- We have described three affected siblings from a non-consanguineous family with Corneal Endothelial Dystrophy 2. PMID: 25138764
- Our observations suggest that congenital hereditary endothelial dystrophy caused by homozygous SLC4A11 mutations progresses to Harboyan syndrome, but the severity of this may vary considerably. PMID: 24351571
- Variation in the COL8A2, SLC4A11, and ZEB1 genes is present in only a small fraction of African American cases and as such does not appear to significantly contribute to the genetic risk of Fuchs endothelial corneal dystrophy. PMID: 24348007
- Data shows that the function of SLCA11 is to facilitate the movement of water across the basolateral corneal epithelium. PMID: 23813972
- To the best of our knowledge, this is the first Korean case of CHED2, confirmed by the c.1239C>A (p.C413*) mutation in the SLC4A11 gene, which has not been previously reported. PMID: 23615275
- SLC4A11 has significant EIPA-sensitive Na(+)-OH(-)(H(+)) and NH4(+) permeability. PMID: 23864606
- The purpose of this study was to identify the genetic cause of congenital hereditary endothelial dystrophy 2 in six Indian families and catalog all known mutations in the SLC4A11 gene. PMID: 23922488
- Both substitution c.214+242C > T in IL1RN and novel deletion c.2558+149_2558+203del54 in SLC4A11 were observed significantly more frequently in family members with keratoconus. PMID: 23462747
- SLC4A11 is necessary for cell survival and may explain the pathologic corneal endothelial cell loss in endotheliopathies due to SLC4A11 mutations. PMID: 22447871
- The reduction in movement of WT SLC4A11 protein to the cell surface caused by Fuchs endothelial corneal dystrophy SLC4A11 helps to explain the dominant inheritance of this disorder. PMID: 22072594
- biochemical study of SLC4A11 PMID: 21288032
- The present study detected one novel and three reported changes, adding to the repertoire of mutations in SLC4A11, and recorded a high degree of genetic heterogeneity in Congenital Hereditary Endothelial Dystrophy. PMID: 21203343
- sequenced SLC4A11 in 192 sporadic and small nuclear late-onset Fuchs corneal dystrophy families and found seven heterozygous missense novel variations that were absent from ethnically matched controls PMID: 20848555
- Corneal endothelial cells are more vulnerable to defects in the functional activity of SLC4A11 than cells of the striae vascularis of the inner ear. PMID: 20118786
- The corneal dystrophy and perceptive deafness (Harboyan syndrome) gene (CDPD1) maps to chromosome 20p13. PMID: 11836359
- describe seven different mutations in the SLC4A11 gene in ten families with autosomal recessive congenital hereditary endothelial dystrophy PMID: 16767101
- These results confirm that mutations in the SLC4A11 gene cause autosomal recessive corneal endothelial dystrophy. PMID: 16825429
- These findings extend the implication of the SLC4A11 borate transporter beyond corneal dystrophy to perceptive deafness. PMID: 17220209
- Novel indel mutation, c.859_862delGAGAinsCCT (E287fsX21) in exon 8 of SLC4A11 gene. Novel in-frame deletion mutation c.2014_2016delTTC or 2017_2019delTTC which will lead to loss of a phenylalanine residue at position 672 or 673 (F672del or F673del). PMID: 17262014
- report of seven novel mutations and two previously identified mutations in families from India and the United Kingdom with recessive congenital hereditary endothelial dystrophy PMID: 17397048
- CHED2 (congenital hereditary endothelial dystrophy) is associated with mutations in SLC4A11, a member of the SLC4 family of base transporters. PMID: 17667634
- These data add to the mutational repertoire of SLC4A11 and establish the high degree of mutational heterogeneity in autosomal recessive congenital hereditary endothelial dystrophy. PMID: 17679935
- SLC4A11 mutations in Fuchs' endothelial corneal dystrophy are reported. PMID: 18024964
- A novel SLC4A11 mutation (Thr271Met) is associated with autosomal recessive congenital hereditary endothelial dystrophy in a pedigree from the Kingdom of Saudi Arabia and provides additional support that mutations in this gene cause disease. PMID: 18363173
- This study increases the number of SLC4A11 gene mutations and confirms the role of this gene in causing congenital hereditary endothelial dystrophy (CHED2). PMID: 18474783
- In this small cohort, no evidence was found of genetic heterogeneity in congenital hereditary endothelial dystrophy (CHED) and that loss of BTR1 function is the most likely mutational mechanism. PMID: 19369245
顯示更多
收起更多
-
相關疾病:Corneal dystrophy and perceptive deafness (CDPD); Corneal endothelial dystrophy (CHED); Corneal dystrophy, Fuchs endothelial, 4 (FECD4)
-
亞細胞定位:Cell membrane; Multi-pass membrane protein. Basolateral cell membrane; Multi-pass membrane protein.
-
蛋白家族:Anion exchanger (TC 2.A.31) family
-
組織特異性:Widely expressed. Highly expressed in kidney, testis, salivary gland, thyroid, trachea and corneal endothelium. Not detected in retina and lymphocytes.; [Isoform 3]: Expressed in corneal endothelium (at protein level).; [Isoform 5]: The predominant isofor
-
數據庫鏈接:
Most popular with customers
-
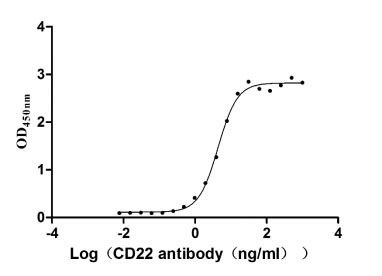
Recombinant Human B-cell receptor CD22 (CD22), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
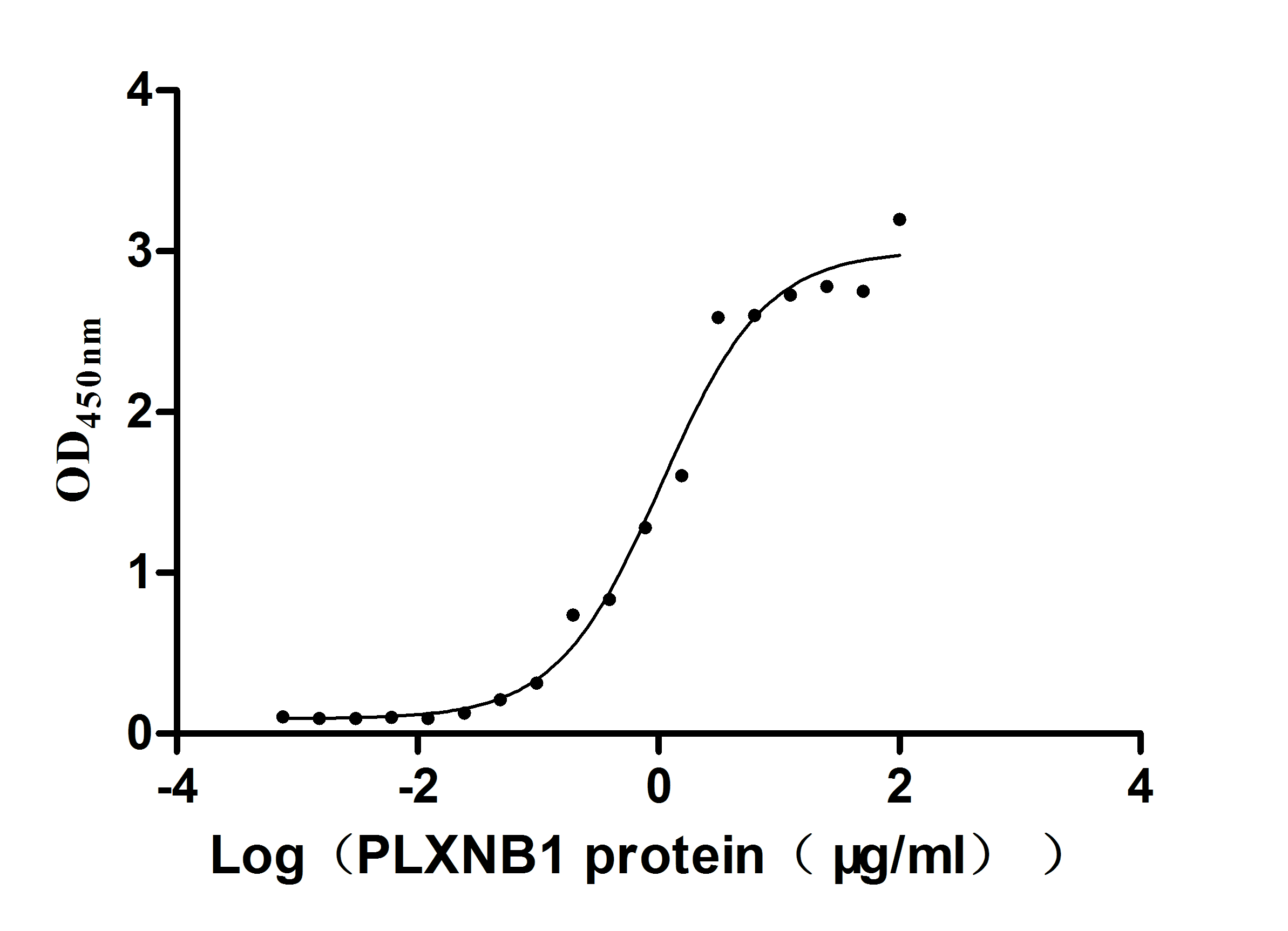
Recombinant Human Plexin-B1 (PLXNB1), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
-AC1.jpg)
Recombinant Human HLA class II histocompatibility antigen gamma chain (CD74), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
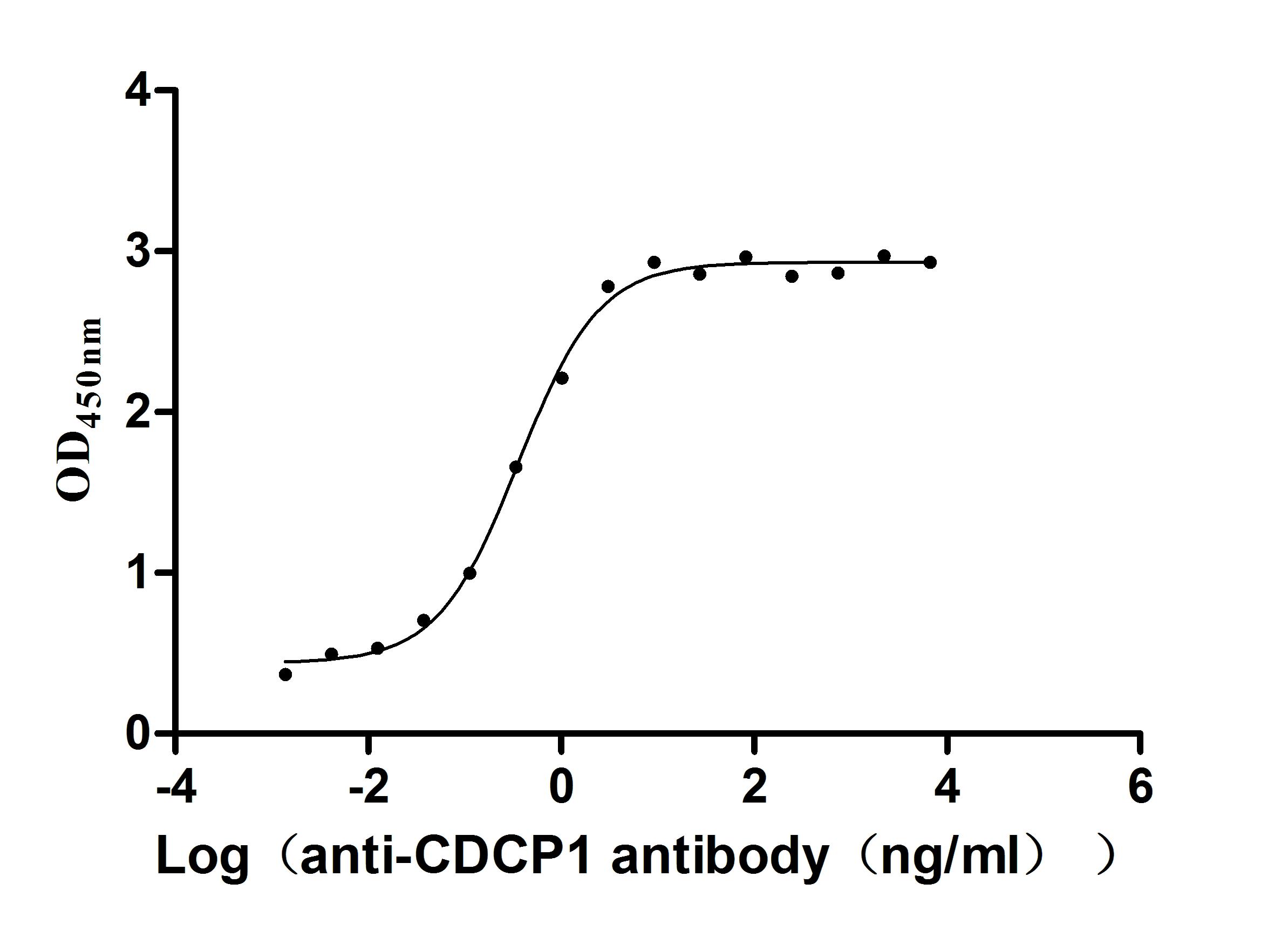
Recombinant Human CUB domain-containing protein 1 (CDCP1), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
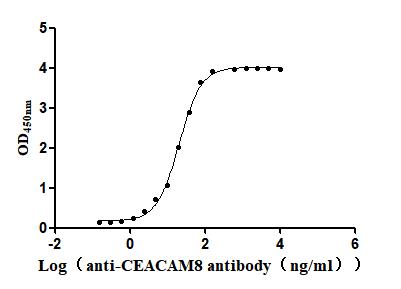
Recombinant Human Carcinoembryonic antigen-related cell adhesion molecule 8(CEACAM8) (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
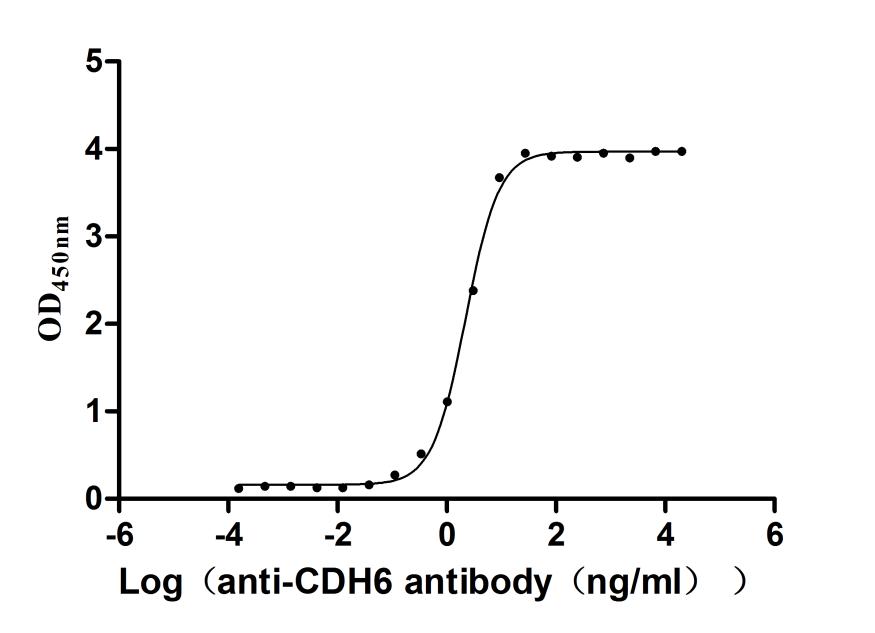
Recombinant Macaca fascicularis Cadherin 6(CDH6),partial (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)









