GAA Recombinant Monoclonal Antibody
-
中文名稱:GAA重組抗體
-
貨號:CSB-RA566370A0HU
-
規(guī)格:¥1320
-
圖片:
-
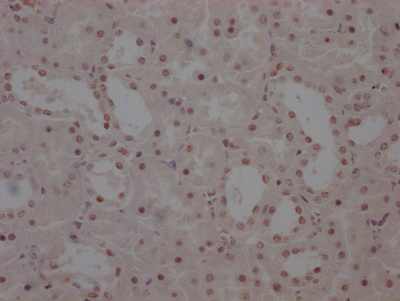
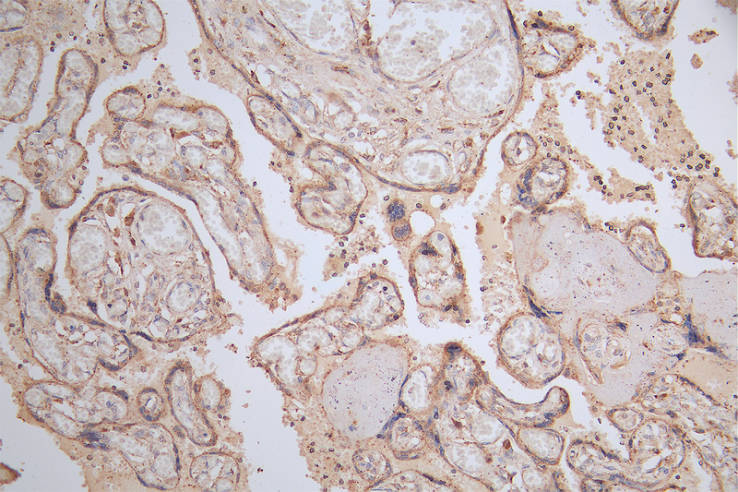 IHC image of CSB-RA566370A0HU diluted at 1:50 and staining in paraffin-embedded human placenta tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.15% DAB.
IHC image of CSB-RA566370A0HU diluted at 1:50 and staining in paraffin-embedded human placenta tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.15% DAB. -
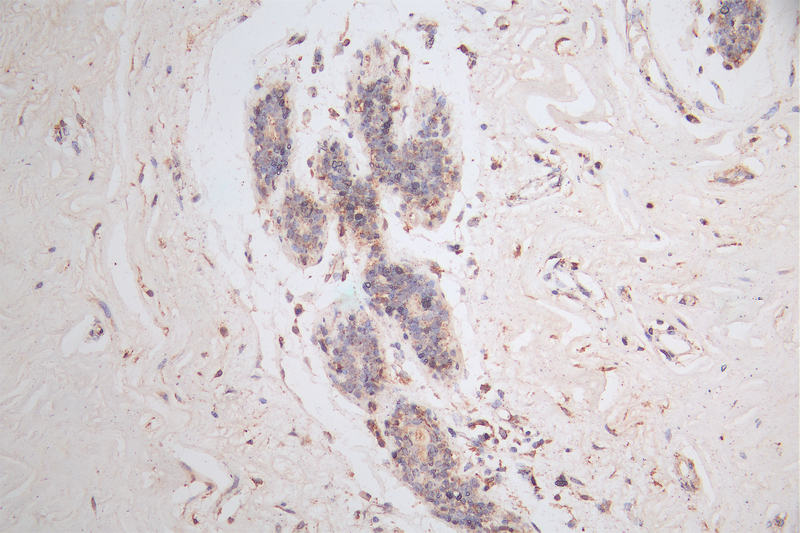 IHC image of CSB-RA566370A0HU diluted at 1:50 and staining in paraffin-embedded human breast cancer performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.15% DAB.
IHC image of CSB-RA566370A0HU diluted at 1:50 and staining in paraffin-embedded human breast cancer performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.15% DAB.
-
-
其他:
產(chǎn)品詳情
-
產(chǎn)品描述:CSB-RA566370A0HU GAA重組單克隆抗體是針對酸性α-葡萄糖苷酶(GAA)靶點開發(fā)的高特異性科研工具,適用于酶聯(lián)免疫吸附(ELISA)和免疫組織化學(xué)(IHC)實驗。該抗體識別的GAA蛋白是溶酶體糖原代謝的關(guān)鍵酶,其功能異常與糖原貯積病II型(龐貝病)等遺傳代謝疾病密切相關(guān)。經(jīng)實驗驗證,本產(chǎn)品在IHC應(yīng)用中展現(xiàn)優(yōu)異的組織抗原定位能力,推薦使用稀釋比例為1:50至1:200,可清晰檢測石蠟包埋樣本中GAA的細胞特異性表達分布。其在ELISA平臺的應(yīng)用則支持靶蛋白的定量分析需求,為研究人員提供可靠的檢測數(shù)據(jù)。該抗體適用于疾病模型構(gòu)建、代謝通路研究及治療性酶活性調(diào)控等基礎(chǔ)科研領(lǐng)域,特別在糖原代謝異常相關(guān)疾病的病理機制探索、基因治療靶點驗證等方向具有應(yīng)用價值。通過嚴格的免疫原篩選和表位驗證,確保抗體與GAA蛋白的高度結(jié)合特異性,為體外實驗提供精準的檢測保障。
-
Uniprot No.:
-
基因名:
-
別名:Lysosomal alpha-glucosidase (EC 3.2.1.20) (Acid maltase) (Aglucosidase alfa) [Cleaved into: 76 kDa lysosomal alpha-glucosidase, 70 kDa lysosomal alpha-glucosidase], GAA
-
反應(yīng)種屬:Human
-
免疫原:A synthesized peptide derived from Human GAA
-
免疫原種屬:Homo sapiens (Human)
-
標記方式:Non-conjugated
-
克隆類型:Monoclonal
-
抗體亞型:Rabbit IgG
-
純化方式:Affinity-chromatography
-
克隆號:5E5
-
濃度:It differs from different batches. Please contact us to confirm it.
-
保存緩沖液:Rabbit IgG in 10mM phosphate buffered saline , pH 7.4, 150mM sodium chloride, 0.05% BSA, 0.02% sodium azide and 50% glycerol.
-
產(chǎn)品提供形式:Liquid
-
應(yīng)用范圍:ELISA, IHC
-
推薦稀釋比:
Application Recommended Dilution IHC 1:50-1:200 -
Protocols:
-
儲存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產(chǎn)品評價

相關(guān)產(chǎn)品
靶點詳情
-
功能:Essential for the degradation of glycogen in lysosomes. Has highest activity on alpha-1,4-linked glycosidic linkages, but can also hydrolyze alpha-1,6-linked glucans.
-
基因功能參考文獻:
- PI-rhGAA may have the potential to be a useful therapeutic option for improving the treatment of Pompe disease. PMID: 29102549
- The most common mutation was c.-32-13T, G. in Pompe disease. PMID: 29181627
- The narrow substrate-binding pocket of rhGAA is located near the C-terminal ends of beta-strands of the catalytic (beta/alpha)8 domain and shaped by a loop from the N-terminal beta-sheet domain and both inserts I and II. PMID: 29061980
- This is the first study of rhGAA to differentiate M6P glycans and identify their attachment sites, despite rhGAA already being an approved drug for Pompe disease. PMID: 29274340
- GAA mutation is associated with Pompe disease. PMID: 28763149
- Enzyme activities (acid alpha-glucosidase (GAA), galactocerebrosidase (GALC), glucocerebrosidase (GBA), alpha-galactosidase A (GLA), alpha-iduronidase (IDUA) and sphingomyeline phosphodiesterase-1 (SMPD-1)) were measured on ~43,000 de-identified dried blood spot (DBS) punches, and screen positive samples were submitted for DNA sequencing to obtain genotype confirmation of disease risk PMID: 27238910
- enzyme replacement therapy (ERT) (alglucosidase alfa) stabilizes respiratory function and improves mobility and muscle strength in late-onset Pompe disease.Lysosomal glycogen in muscle biopsies from treatment-naive LOPD patients was reduced post-ERT (alglucosidase alfa). PMID: 27473031
- In adults with Pompe disease, antibody formation does not interfere with rhGAA efficacy in the majority of patients, is associated with IARs, and may be attenuated by the IVS1/delex18 GAA genotype PMID: 27362911
- Reanalysis of the patient's DNA sample using next generation sequencing (NGS) of a panel of target genes causing glycogen storage disorders demonstrated compound heterozygosity for a point mutation and an exonic deletion in the GAA gene. PMID: 28657663
- Thirteen novel and two common GAA mutations were identified in this study. The allelic frequency of c.2662G > T (p.Glu888X) was 23.1% in northern Chinese patients and 4.2% in southern Chinese patients, whereas the allelic frequency of c.1935C >A (p.Asp645Glu) was 20.8% in southern and 3.8% in northern Chinese patients. PMID: 28394184
- This is the first report of the alpha-glucosidase inhibitory activity of compounds 20, 26, and 29, and the findings support the important role of Eremanthus species as novel sources of new drugs and/or herbal remedies for treatment of type 2 diabetes. PMID: 27322221
- Compared with controls, GAA gene expression levels in coronary artery disease (CAD) patients were significantly increased, suggesting that GAA may be involved in the CAD development. PMID: 26580301
- Study reports on the clinical, biochemical, morphological, muscle imaging, and genetic findings of six adult Pompe patients from five unrelated families with the c.-32-13T>G GAA gene mutation in homozygous state. All patients had decreased GAA activity and elevated creatine kinase levels. PMID: 26231297
- glycogen storage disease type II is caused by deficiency of GAA activity resulting from mutation of GAA gene PMID: 26575883
- RT-PCR followed by DNA sequence analysis of patients with Pompe disease revealed new variant in GAA gene resulting in aberrant splicing event. PMID: 25243733
- Findings indicate that GAA c.2238G > C (p.W746C) novel mutation is the most common mutation in mainland Chinese late-onset Pompe patients, as observed in Taiwanese patients expanding the genetic spectrum of the disease. PMID: 25526786
- this study shows several alterations distributed along the GAA gene in a sample of Brazilian families. PMID: 25681614
- Mutations in acid alpha-glucosidase gene is associated with Pompe disease. PMID: 25026126
- GAA deficiency results in reduced mTORC1 activation that is partly responsible for the skeletal muscle wasting phenotype and can be amerliorated by leucine supplementation. PMID: 25231351
- The phenotype LO-GSDII with GAA mutation in the North of Italy seems not significantly different from other LO-GSDII populations in Europe or the USA. PMID: 24158270
- Data shows the largest informative family with late-onset Pompe disease described in the literature showing a peculiar complex set of mutations of GAA gene that may partially elucidate the clinical heterogeneity of this family. PMID: 24107549
- 7 of 27 in: Gene. 2014 Mar 1;537(1) Novel GAA sequence variant c.1211 A>G reduces enzyme activity but not protein expression in infantile and adult onset Pompe disease. PMID: 24384324
- This study demonstrates that the c.-32-13T>G mutation of GAA gene abrogates the binding of the splicing factor U2AF65 to the polypyrimidine tract of exon 2 and that several splicing factors affect exon 2 inclusion. PMID: 24150945
- study describes two unrelated cases affected with classical early-onset Pompe disease, both pertaining to the same small Mexican region, with the same novel homozygous frameshift mutation at gene GAA (c.1987delC) PMID: 24399866
- Mutations in the GAA gene is associated with glycogen storage disease type II. PMID: 23884227
- Adult patients with alpha-glucosidase mutations other than c.-32-13 T>G can have very low alpha-glucosidase activity in fibroblasts but express higher activity in muscle and store less glycogen in muscle than patients with infantile Pompe disease. PMID: 23000108
- Study gave an update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. PMID: 22644586
- Transcriptional response to GAA deficiency (Pompe disease) in infantile-onset patients PMID: 22658377
- Report genetic testing to indentify GAA mutations in German patients with late-onset glycogen storage disease type II. PMID: 18607768
- we define a critical role for endoplasmic reticulum stress in the activation of autophagy due to the 546G>T acid alpha glucosidase mutation PMID: 21982629
- No common mutation is found in association with low levels of acid alpha-glucosidase activity in late-onset Pompe disease; most patients produce unprocessed forms of GAA protein compared with patients who have higher GAA activity. PMID: 21484825
- Mutation analysis of the GAA gene revealed the p.D645E in all patients with Pompe disease, suggesting it as the most common mutation in the Thai population. PMID: 21039225
- The enzymatic screening of Pompe disease can be justified in patients with myopathies of unknown etiology in this report of a Mexican patient with late-onset glycogen-storage disease type 2. PMID: 20350966
- Data show that p.R1147G missense mutation impaired glucosidase activity. PMID: 19834502
- Homozygosity for multiple contiguous single-nucleotide polymorphisms as an indicator of large heterozygous deletions: identification of a novel heterozygous 8-kb intragenic deletion (IVS7-19 to IVS15-17) in a patient with glycogen storage disease type II PMID: 11854868
- novel target of the Notch-1/Hes-1 signaling pathway PMID: 12065598
- 2 novel mutations of the acid alpha-glucosidase gene, P361L and R437C, were found in a juvenile-onset glycogen storage disease type II (GSDII) 16-year-old Chinese patient. The asymptomatic 13-year-old brother of the proband is also compound heterozygote PMID: 12601120
- mutations in the alpha glucosidase gene is associated with infantile onset glycogen storage disease type II. PMID: 12923862
- Childhood Pompe disease demonstrating phenotypic variability of p.Asp645Asn. PMID: 15145338
- data show that the mature forms of GAA characterized by polypeptides of 76 or 70 kDa are in fact larger molecular mass multicomponent enzyme complexes; peptides released during proteolytic processing remained tightly associated with the major species PMID: 15520017
- 2 novel mutations (Ala237Val and Gly293Arg) were foundin the acid alpha-glucosidase gene in a Pompe disease patient with vascular involvement. PMID: 15668445
- Acid-alpha-glucosidase activity and specific activity, and lysosomal glycogen content are useful predictors of age of onset in Pompe disease PMID: 15993875
- Complete molecular analysis of the GAA gene of patients with late onset glycogen storage disease type II shows missense mutations and splicing mutations. PMID: 16917947
- From 14 Argentinean patients diagnosed with either infantile or late-onset disease, we identified 14 distinct mutations in the acid alpha-glucosidase (GAA) gene including nine novel variants. PMID: 17056254
- Two new missense mutations (p.266Pro>Ser and p.439Met>Lys) were new missense mutations causing late onset GSD II. PMID: 17092519
- Patients with the same c.-32-13T-->G haplotype (c.q. GAA genotype) may manifest first symptoms at different ages, indicating that secondary factors may substantially influence the clinical course of patients with this mutation. PMID: 17210890
- demonstrated a significant increase of GAA activity (1.3-7.5-fold) after imino sugar treatment in fibroblasts from patients carrying the mutations L552P (three patients) and G549R (one patient) PMID: 17213836
- N-glycans of recombinant human GAA were expressed in the milk of transgenic rabbits. PMID: 17293352
- The role of autophagy in Pompe disease was examined by analyzing single muscle fibers. PMID: 17592248
- Mutations in glucosidase alpha is asspciated with glycogen storage disease type II PMID: 17616415
顯示更多
收起更多
-
相關(guān)疾病:Glycogen storage disease 2 (GSD2)
-
亞細胞定位:Lysosome. Lysosome membrane.
-
蛋白家族:Glycosyl hydrolase 31 family
-
數(shù)據(jù)庫鏈接:
Most popular with customers
-
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-
-