AGL Recombinant Monoclonal Antibody
-
中文名稱:AGL Recombinant Monoclonal Antibody
-
貨號:CSB-RA116104A0HU
-
規格:¥1320
-
圖片:
-
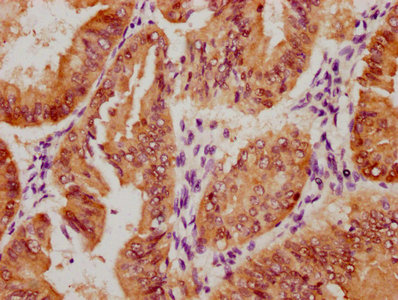
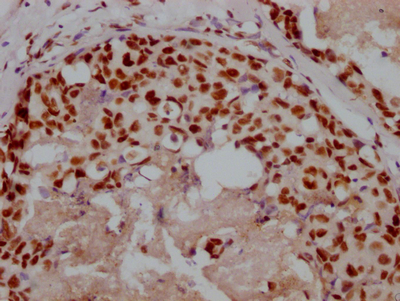
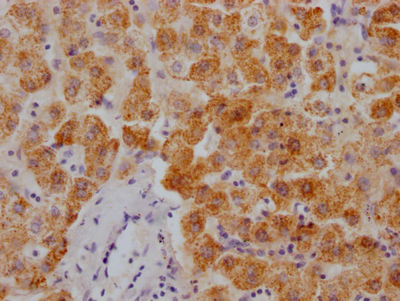
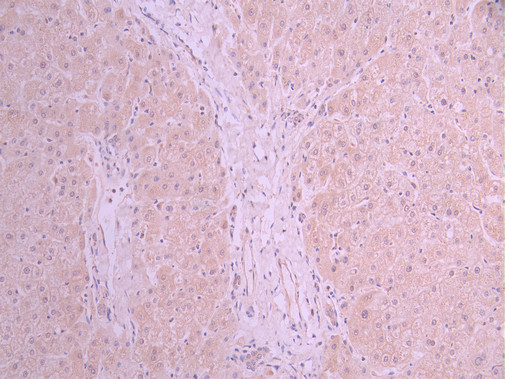 IHC image of CSB-RA116104A0HU diluted at 1:100 and staining in paraffin-embedded human liver tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB.
IHC image of CSB-RA116104A0HU diluted at 1:100 and staining in paraffin-embedded human liver tissue performed on a Leica BondTM system. After dewaxing and hydration, antigen retrieval was mediated by high pressure in a citrate buffer (pH 6.0). Section was blocked with 10% normal goat serum 30min at RT. Then primary antibody (1% BSA) was incubated at 4°C overnight. The primary is detected by a Goat anti-rabbit polymer IgG labeled by HRP and visualized using 0.05% DAB. -
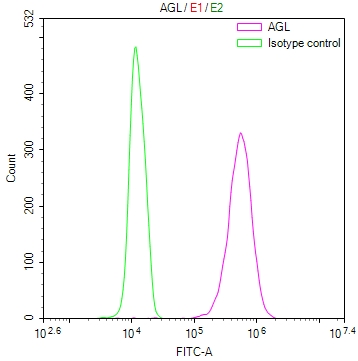 Overlay Peak curve showing Hela cells stained with CSB-RA116104A0HU (red line) at 1:100. The cells were fixed in 4% formaldehyde and permeated by 0.2% TritonX-100 for?10min. Then 10% normal goat serum to block non-specific protein-protein interactions followed by the antibody (1ug/1*106cells) for 45min at 4℃. The secondary antibody used was FITC-conjugated goat anti-rabbit IgG (H+L) at 1/200 dilution for 35min at 4℃.Control antibody (green line) was Rabbit IgG (1ug/1*106cells) used under the same conditions. Acquisition of >10,000 events was performed.
Overlay Peak curve showing Hela cells stained with CSB-RA116104A0HU (red line) at 1:100. The cells were fixed in 4% formaldehyde and permeated by 0.2% TritonX-100 for?10min. Then 10% normal goat serum to block non-specific protein-protein interactions followed by the antibody (1ug/1*106cells) for 45min at 4℃. The secondary antibody used was FITC-conjugated goat anti-rabbit IgG (H+L) at 1/200 dilution for 35min at 4℃.Control antibody (green line) was Rabbit IgG (1ug/1*106cells) used under the same conditions. Acquisition of >10,000 events was performed.
-
-
其他:
產品詳情
-
Uniprot No.:
-
基因名:AGL
-
別名:Glycogen debranching enzyme (Glycogen debrancher) [Includes: 4-alpha-glucanotransferase (EC 2.4.1.25) (Oligo-1,4-1,4-glucantransferase), Amylo-alpha-1,6-glucosidase (Amylo-1,6-glucosidase) (EC 3.2.1.33) (Dextrin 6-alpha-D-glucosidase)], AGL, GDE
-
反應種屬:Human
-
免疫原:A synthesized peptide from human AGL protein
-
免疫原種屬:Homo sapiens (Human)
-
標記方式:Non-conjugated
-
克隆類型:Monoclonal
-
抗體亞型:Rabbit IgG
-
純化方式:Affinity-chromatography
-
克隆號:8D4
-
濃度:It differs from different batches. Please contact us to confirm it.
-
保存緩沖液:Preservative: 0.03% Proclin 300
Constituents: 50% Glycerol, 0.01M PBS, PH 7.4 -
產品提供形式:Liquid
-
應用范圍:ELISA, IHC, FC
-
推薦稀釋比:
Application Recommended Dilution IHC 1:50-1:200 FC 1:50-1:200 -
Protocols:
-
儲存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產品評價

相關產品
靶點詳情
-
功能:Multifunctional enzyme acting as 1,4-alpha-D-glucan:1,4-alpha-D-glucan 4-alpha-D-glycosyltransferase and amylo-1,6-glucosidase in glycogen degradation.
-
基因功能參考文獻:
- This report of patients with GSD-III in Iran with 2 uncommon clinical presentations and 5 novel mutations in the AGL gene. PMID: 29794575
- The study identified 31 novel mutations and extended the mutation spectrum of AGL in Chinese patients with glycogen storage disease type III. PMID: 26984562
- Our study establishes HAS2-mediated HA synthesis as a driver of growth of bladder cancer with low AGL and provides preclinical rationale for personalized targeting of HAS2/HA signaling in patients with low amylo-alpha-1-6-glucosidase-4-alpha-glucanotransferase -expressing tumors. PMID: 26490312
- AGL loss causes high SHMT2 expression and consequently increases glycine-dependent nucleotide synthesis leading to bladder cancer growth. PMID: 26975021
- Point mutations in AGL gene are associated with glycogen storage disease type IIIa in a Chinese family. PMID: 26252094
- Haplotype analysis revealed that the mutation arises as a result of founder effect, not an independent event. PMID: 25827695
- AGL haplotype analyses suggested that c.1019delA and c.958+1G>A are founder mutations in Turkish patients, while p.R864X is a recurrent mutation. PMID: 25451950
- A homozygous frameshift deletion, c.4456delT, in exon 33 of the AGL gene in Inuit children determines the cause of glycogen storage disease type IIIa and confirms a founder effect. PMID: 25602008
- study identified 10 different mutations in 8 Korean Glycogen storage disease type III patients; 5 mutations are novel and include 1 nonsense (c.1461G>A, p.W487X), 3 splicing (c.293+4_293+6delAGT in IVS4, c.460+1G>T in IVS5, c.2682-8A>G in IVS21) and 1 missense mutation (c.2591G>C, p.R864P) PMID: 24257475
- Characterization of a novel homozygous single point mutation at the polypyrimidine tract of intron 21 of the AGL gene in two consanguineous siblings with glycogen storage disease type III. PMID: 23649758
- We found that most patients with macular telangiectasia-2 possess retinal autoantibodies, the most prevalent of which were directed against AGL, RBP3, and CK-B. PMID: 23882694
- A founder effect discovered amongst Tunisian patients with glycogen storage disease type III and a c.3216_3217delGA mutation in the AGL gene. PMID: 22035446
- Mutations in amylo-1,6-glucosidase is associated with Glycogen Storage Disease Type III. PMID: 20648714
- The present patient was found to be deficient in GDE activity and homozygous for a novel 1 bp deletion in AGL. This mutation is predicted to cause premature termination at codon 834 due to frame shift. PMID: 20158661
- GSD-III patients have variable phenotypic characteristics. Administration of raw-corn-starch can effectively improve the disease outcome. We identified 8 new mutations on AGL gene through nucleotide sequence analysis. PMID: 15833157
- Nine AGL mutations: six nonsense mutations , one deletion and two splicing mutation were identified in Turkish GSD III patients. PMID: 19834502
- Mutations associated with GSD III include R34X and Y1148X. PMID: 11924557
- it is likely that the AMPK-GDE association is a novel mechanism regulating AMPK activity and the resultant fatty acid oxidation and glucose uptake PMID: 15886229
- AGL gene mutations may have roles in glycogen storage disease type III PMID: 17047887
- These results indicate that binding to glycogen crucially regulates the stability of AGL and, further, that its ubiquitination may play an important role in the pathophysiology of both Lafora and Cori's disease. PMID: 17908927
- Current clinical and molecular knowledge about glycogenosis 3 and phenotype and genotype levels of this enzyme. [REVIEW] PMID: 17915576
- a homozygous p.W1327X mutation leads to severe generalized glycogenosis types 3a & 3b within the same family; heterozygous p.W1327X mutation carriers may present with mild non-progressive neuromuscular symptoms, such as exercise-induced myalgia & fatigue PMID: 18924225
- Mutations in the carbohydrate-binding domain of AGL lead to loss of all enzymatic activities and enhancing targeting for proteasomal degradation. PMID: 19299494
- Six novel AGL mutations were identified. PMID: 19754354
顯示更多
收起更多
-
相關疾病:Glycogen storage disease 3 (GSD3)
-
亞細胞定位:Cytoplasm. Note=Under glycogenolytic conditions localizes to the nucleus.
-
蛋白家族:Glycogen debranching enzyme family
-
組織特異性:Liver, kidney and lymphoblastoid cells express predominantly isoform 1; whereas muscle and heart express not only isoform 1, but also muscle-specific isoform mRNAs (isoforms 2, 3 and 4). Isoforms 5 and 6 are present in both liver and muscle.
-
數據庫鏈接:
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-