SPAST Antibody
-
中文名稱:SPAST兔多克隆抗體
-
貨號(hào):CSB-PA587093
-
規(guī)格:¥1100
-
圖片:
-
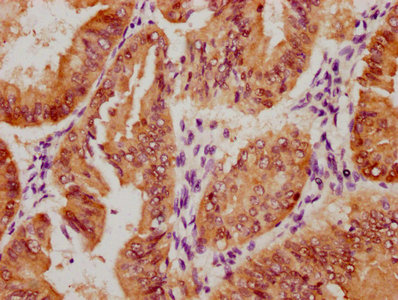
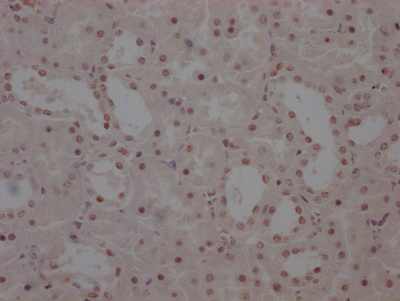
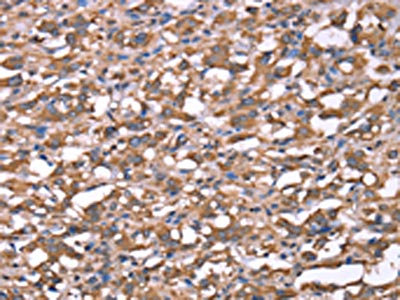 The image on the left is immunohistochemistry of paraffin-embedded Human thyroid cancer tissue using CSB-PA587093(SPAST Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human thyroid cancer tissue using CSB-PA587093(SPAST Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200) -
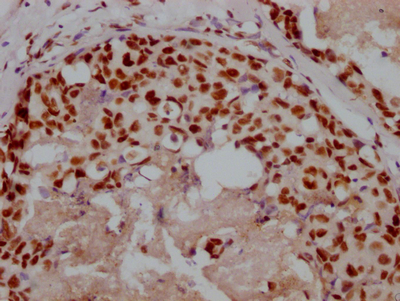
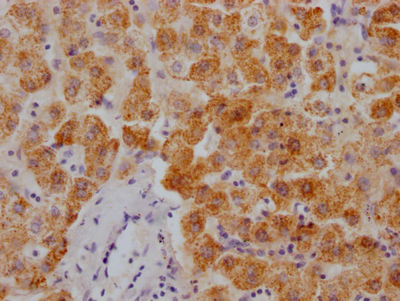
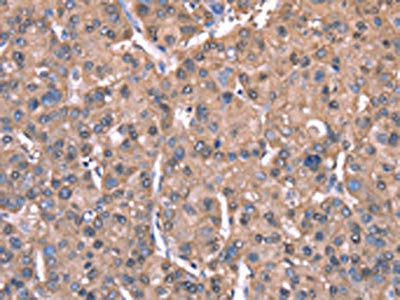 The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using CSB-PA587093(SPAST Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using CSB-PA587093(SPAST Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200)
-
-
其他:
產(chǎn)品詳情
-
Uniprot No.:
-
基因名:SPAST
-
別名:SPAST antibody; ADPSP antibody; FSP2 antibody; KIAA1083 antibody; SPG4 antibody; Spastin antibody; EC 5.6.1.1 antibody; Spastic paraplegia 4 protein antibody
-
宿主:Rabbit
-
反應(yīng)種屬:Human,Mouse,Rat
-
免疫原:Synthetic peptide of Human SPAST
-
免疫原種屬:Homo sapiens (Human)
-
標(biāo)記方式:Non-conjugated
-
抗體亞型:IgG
-
純化方式:Antigen affinity purification
-
濃度:It differs from different batches. Please contact us to confirm it.
-
保存緩沖液:-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
-
產(chǎn)品提供形式:Liquid
-
應(yīng)用范圍:ELISA,IHC
-
推薦稀釋比:
Application Recommended Dilution ELISA 1:2000-1:5000 IHC 1:50-1:200 -
Protocols:
-
儲(chǔ)存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產(chǎn)品評(píng)價(jià)

相關(guān)產(chǎn)品
靶點(diǎn)詳情
-
功能:ATP-dependent microtubule severing protein that specifically recognizes and cuts microtubules that are polyglutamylated. Preferentially recognizes and acts on microtubules decorated with short polyglutamate tails: severing activity increases as the number of glutamates per tubulin rises from one to eight, but decreases beyond this glutamylation threshold. Severing activity is not dependent on tubulin acetylation or detyrosination. Microtubule severing promotes reorganization of cellular microtubule arrays and the release of microtubules from the centrosome following nucleation. It is critical for the biogenesis and maintenance of complex microtubule arrays in axons, spindles and cilia. SPAST is involved in abscission step of cytokinesis and nuclear envelope reassembly during anaphase in cooperation with the ESCRT-III complex. Recruited at the midbody, probably by IST1, and participates in membrane fission during abscission together with the ESCRT-III complex. Recruited to the nuclear membrane by IST1 and mediates microtubule severing, promoting nuclear envelope sealing and mitotic spindle disassembly during late anaphase. Required for membrane traffic from the endoplasmic reticulum (ER) to the Golgi and endosome recycling. Recruited by IST1 to endosomes and regulates early endosomal tubulation and recycling by mediating microtubule severing. Probably plays a role in axon growth and the formation of axonal branches.; Involved in lipid metabolism by regulating the size and distribution of lipid droplets.
-
基因功能參考文獻(xiàn):
- The findings suggest a mechanism for neurodegeneration in hereditary spastic paraplegia whereby SPAST mutations indirectly lead to impaired peroxisome transport and oxidative stress. PMID: 27229699
- study to explore the novel SPAST splice site donor variant, c.1004+3A>C in spastic paraplegia type 4; Exon 6 is skipped out by the variant, leading to premature termination of translation, p.Gly290Trpfs*5; measurement of SPAST transcripts in lymphocytes demonstrated reduction through nonsense-mediated mRNA decay PMID: 28870597
- Two novel mutations in gene SPG4 in patients with autosomal dominant spastic paraplegia PMID: 29368828
- The N184X mutation triggers the reinitiation of translation at a third start codon in SPAST, resulting in synthesis of a novel M187 spastin isoform that is able to sever microtubules. PMID: 28495799
- Our data reveal a high rate of complex cases (25%), with psychiatric disorders among the most common comorbidity (10% of all SPASTpatients). Further, we identify a genotype-phenotype correlation between patients carrying loss-of-function mutations in SPAST and the presence of psychiatric disorders. PMID: 28572275
- Variants in SPAST and KIF5A were the most common causes of autosomal dominant hereditary spastic paraplegia in Greece. PMID: 26374131
- Two distinct Alu insertion-associated deletions in the SPAST gene cause hereditary spastic paraplegia type SPG4. PMID: 26932189
- The data of this study confirmed the genetic heterogeneity of childhood-onset pure HSP, with SPG4/SPAST and SPG3A/ATL1 being the most frequent forms. PMID: 27260292
- We report the first genetic study of uncomplicated HSP patients from the Czech Republic. We found broad mutation spectrum in 13 from the 17 coding exons and adjacent regions of the SPAST gene. We detected 21 novel presumably pathogenic mutations. The high frequency of SPAST mutations was found only in familial patients. PMID: 27334366
- This study demonstrted that the most frequently affected gene was SPAST with pathogenic or likely pathogenic mutations in hereditary spastic paraplegia. PMID: 27084228
- This study demonstrated that SPAST gene mutation associated with hereditary spastic paraplegias in group of Polish patients PMID: 26671083
- We identified two novel mutations and two previously reported mutations in SPAST and ATL1, respectively. The family with the ATL1 c.1204T>G mutation exhibited male-lethality, female infancy-onset, and pseudo- X-linked dominant transmission PMID: 26600529
- Using human-engineered and differentially modified microtubules study finds that glutamylation is the main regulator of the hereditary spastic paraplegia microtubule severing enzyme spastin. PMID: 26875866
- Patients with deletions of exons in the SPAST gene showed pure hereditary spastic paraplegia. PMID: 26165777
- Novel SPAST pathogenic variants were identified in Korean patients with hereditary spastic paraplegia. PMID: 26208798
- The coexistence of mutations in SPAST and FSHD was confirmed in our proband and in two siblings PMID: 25511172
- The spastin as a microtubule-severing protein was an important mechanistic breakthrough, it seems certain that insufficient microtubule severing alone is not an adequate explanation for HSP-SPG4. PMID: 26094131
- SPAST mutations are common in Chinese patients with pure hereditary spastic paraplegia PMID: 24824479
- Data showed 3 micro-mutations and 2 exon deletions in SPAST gene and 2 micro-mutations in ATL1 gene in this cohort of Chinese patients with spastic paraplegia. PMID: 25454648
- NA14 may act as an adaptor protein regulating SPG4 localization to centrosomes, temporally and spatially regulating the microtubule-severing activity of SPG4 that is particularly critical during the cell cycle and neuronal development. PMID: 25390646
- ESCRT-III, VPS4 and spastin cooperate to coordinate nuclear envelope sealing and spindle disassembly at nuclear envelope-microtubule intersection sites during mitotic exit to ensure nuclear integrity and genome safeguarding PMID: 26040712
- SPG4 appears to be the major cause of hereditary spastic paraplegia in Tuscany. PMID: 24731568
- This study reported a novel splice-site mutation, c.1098+1~2GT-->CTCAGA, in the AAA domain of SPG4 in a Chinese family with pure autosomal dominant hereditary spastic paraplegia. PMID: 24659419
- four novel mutations were identifiedin SPAST/SPG4. PMID: 24033003
- analys of deletion mutation of SPAST in hereditary spastic paraplegias, correlation with increased tendon reflexes in the lower limbs and Babinski sign PMID: 24824741
- Neurite complexity and maintenance in hereditary spastic paraplegia patient-derived neurons are critically sensitive to spastin gene dosage. PMID: 24381312
- the Alu genomic architecture of SPAST predisposes to diverse intragenic copy-number variants alleles with distinct transcriptional--and possibly phenotypic--consequences. PMID: 25065914
- successful establishment of human pluripotent stem cell-based neuronal models of SPG4, which will be valuable for dissecting the pathogenic cellular mechanisms and screening compounds to rescue the axonal degeneration in hereditary spastic paraplegias PMID: 24123785
- This study identified the genetic cause in approximately 25 % of patients in this sample; this is a high proportion of cases, given that one of the most common causes of HSP (SPG4) had already been excluded. PMID: 23812641
- Our study enlarges the number of pathogenic SPAST mutations, and confirms the association with a pure spastic paraplegia phenotype PMID: 22960362
- This study demonistrated that toxicity of mutant spastin proteins, especially mutant M1, contributes to axonal degeneration in the corticospinal tracts. PMID: 24478365
- a novel 14-bp heterozygous deletion that induced a frameshift mutation in exon 15 of SPAST is predicted to have functional impact and found to cosegregate with the disease phenotype of hereditary spastic paraplegia PMID: 23716148
- The results suggest that inclusion of IST1 into the ESCRT complex allows recruitment of spastin to promote fission of recycling tubules from the endosome. PMID: 23897888
- Data suggest that ATP-bound SPG4 interacts strongly/cooperatively with microtubules; this interaction stimulates ATP hydrolysis by SPG4; SPG4 then dissociates from microtubules and exchanges ADP for ATP in solution for next round/cycle. PMID: 23745751
- Patients with SPG4-related hereditary spastic paraplegia are not found to have ophthalmological manifestations. PMID: 23238845
- Compared to control cells, patient-derived cells had 50% spastin, 50% acetylated alpha-tubulin and 150% stathmin, a microtubule-destabilizing enzyme. PMID: 23264559
- micro-rearrangements in the SPAST gene are a fairly frequent cause of hereditary spastic paraplaegia PMID: 22203332
- study concludes that SPAST mutations are responsible for the majority of hereditary spastic paraplegia (HSP) in Australia; most of the patients with SPAST mutations had pure forms of HSP and a positive family history to suggest autosomal dominant HSP PMID: 23252998
- analysis of spastin's microtubule-binding properties and comparison with katanin PMID: 23272056
- The results of this study a a SPG4 mutation was higher than for patients with SPG3 mutations in patients with Autosomal dominant spastic paraplegias. PMID: 23400676
- Data report here the 3.3 A X-ray crystal structure the AAA domain of human spastin (SPG4) and show that, despite amino acid differences in a number of key residues, the human and D. melanogaster spastin structures are highly conserved. PMID: 22446388
- findings indicate that protrudin interacts with spastin and induces axon formation through its N-terminal domain; protrudin and spastin may work together to play an indispensable role in motor axon outgrowth PMID: 22573551
- wild type spastin is even more sensitive toward the presence of inactive mutants than in enzymatic assays, suggesting a weak coupling of ATPase and severing activity. PMID: 22637577
- transcriptional and post-transcriptional regulation of SPAST PMID: 22574173
- Peripheral neuropathy occurs in hereditary spastic paraplegia patients with SPG4 mutations. PMID: 22192498
- We identified seven different spastin mutations in five probands and one sporadic patient with Hereditary spastic paraplegia PMID: 21834905
- Spastin was identified as a novel component of the HOXA10 transcriptional complex in Ishikawa nuclear extracts. PMID: 21757506
- The results of this study showed that consistent with data suggesting that SPAST mutations mostly cause a pure HSP phenotype. PMID: 21546041
- previously unreported autosomal dominant mutations in the spastin gene in hereditary spastic paraplegia PMID: 20718791
- A higher level (78.8 +/- 3.9%) of functional spastin than the expected ratio of 50% owing to leaky splicing might cause late age at onset of hereditary spastic paraplegia. PMID: 20491894
顯示更多
收起更多
-
相關(guān)疾?。?/div>Spastic paraplegia 4, autosomal dominant (SPG4)亞細(xì)胞定位:Membrane; Peripheral membrane protein. Endoplasmic reticulum. Midbody. Cytoplasm, cytoskeleton, microtubule organizing center, centrosome. Cytoplasm, cytoskeleton. Cytoplasm, perinuclear region. Nucleus. Cytoplasm, cytoskeleton, spindle. Cytoplasm.; [Isoform 1]: Endoplasmic reticulum membrane; Peripheral membrane protein. Nucleus membrane. Lipid droplet. Cytoplasm, cytoskeleton. Endosome.; [Isoform 3]: Cytoplasm. Endosome. Nucleus membrane.蛋白家族:AAA ATPase family, Spastin subfamily組織特異性:Expressed in brain, heart, kidney, liver, lung, pancreas, placenta and skeletal muscle. The short isoforms may predominate in brain and spinal cord.數(shù)據(jù)庫鏈接:
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-