PMM2 Antibody
-
中文名稱:PMM2兔多克隆抗體
-
貨號(hào):CSB-PA018238GA01HU
-
規(guī)格:¥3,900
-
其他:
產(chǎn)品詳情
-
Uniprot No.:
-
基因名:PMM2
-
別名:AI585868 antibody; BOS_22465 antibody; C86848 antibody; CDG 1 antibody; CDG1 antibody; CDG1a antibody; CDGS antibody; MGC127449 antibody; Phosphomannomutase 2 antibody; PMM 2 antibody; Pmm2 antibody; PMM2_HUMAN antibody
-
宿主:Rabbit
-
反應(yīng)種屬:Human,Mouse,Rat
-
免疫原:Human PMM2
-
免疫原種屬:Homo sapiens (Human)
-
抗體亞型:IgG
-
純化方式:Antigen Affinity Purified
-
濃度:It differs from different batches. Please contact us to confirm it.
-
保存緩沖液:PBS with 0.1% Sodium Azide, 50% Glycerol, pH 7.3. -20°C, Avoid freeze / thaw cycles.
-
產(chǎn)品提供形式:Liquid
-
應(yīng)用范圍:ELISA,WB,IHC
-
Protocols:
-
儲(chǔ)存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產(chǎn)品評(píng)價(jià)

相關(guān)產(chǎn)品
靶點(diǎn)詳情
-
功能:Involved in the synthesis of the GDP-mannose and dolichol-phosphate-mannose required for a number of critical mannosyl transfer reactions.
-
基因功能參考文獻(xiàn):
- The biochemical analysis of mutants does not allow a precise genotypephenotype correlation for PMM2-Type I disorders of glycosylation. PMM2 is very tolerant to missense and loss of function mutations, suggesting that a partial deficiency of activity might be beneficial under certain circumstances. PMID: 30061496
- clinical variables to detect risk factors for Stroke-like episodes in a series of 43 phosphomannomutase deficiency patients. PMID: 29470411
- PMM2-CDG clinical phenotype is heterogeneous in terms of clinical course, with no clear division between neurological and visceral presentations. PMID: 28954837
- Four patients were diagnosed with PMM2-CDG at the age of 8 years or later as their neurological symptoms were quite mild and they had been able to participate in regular school programs. We report patients with p.Val231Met/p.Arg239Trp and p.Ile120Thr/p.Gly228Cys genotypes which may cause milder variants of PMM2-CDG PMID: 28425223
- This functional mouse model of PMM2-CDG, in vitro assays and identification of the novel gp130 biomarker all shed light on the human disease, and moreover, provide the essential tools to test potential therapeutics for this untreatable disease. PMID: 27053713
- We propose that the PMM2 promoter mutation alters tissue-specific chromatin loop formation, with consequent organ-specific deficiency of PMM2 leading to the restricted phenotype of HIPKD. Our findings extend the spectrum of genetic causes for both HI and PKD and provide insights into gene regulation and PMM2 pleiotropy PMID: 28373276
- work describes the functional analysis of 9 PMM2 mutant proteins frequently found in congenital disorder of glycosylation type Ia(PMM2-CDG)patients; results suggest that some loss-of-function mutations detected in PMM2-CDG patients could be destabilizing PMID: 26014514
- the activity of phosphomannomutase2 R141H/F119L heterodimers in vitro, which reproduces the protein found in patients, has the same activity of wild type/R141H, which reproduces the protein found in healthy carriers. PMID: 26488408
- A mild neurological phenotype of PMM2-CDG marked by preserved ambulatory ability and autonomy and associated with L32R mutation is particularly frequent in Italy. PMID: 25355454
- conformational response to ligand binding in phosphomannomutase2 PMID: 25324542
- Data indicate genome-wide significant association at multiple single nucleotide polymorphism (SNPs) near ATP binding cassette transporter 1 (ABCA1) at 9q31.1 and suggestive evidence of association in phosphomannomutase 2 (PMM2) at 16p13.2. PMID: 25173107
- We conclude that electroretinogram signs of on-pathway dysfunction can be detected in the early stages of PMM2-Congenital disorder of glycosylation. PMID: 23430200
- Two young sisters are compound heterozygous for mutations p.Leu32Arg and p.Arg141His, while two paternal great-aunts are compound heterozygosity for p.Leu32Arg and p.Thr237Met, with congenital disorder of glycosylation. PMID: 23988505
- Hypertrophic cardiomyopathy with cardiac rupture and tamponade caused by congenital disorder of glycosylation type Ia with PMM2 mutations in two siblings. PMID: 22374380
- Identification of exclusively catalytic protein change, catalytic protein changes affecting protein stability, two protein changes disrupting the dimer interface and several misfolding changes . PMID: 21541725
- When non-immune hydrops fetalis remains unexplained despite exhaustive obstetrical screening, analysis of PMM activity in the parents' leucocytes is possible and might be performed easily during pregnancy PMID: 20638314
- The presence of this deletion-insertion mutation at cDNA position 565 suggests that this site in the PMM2 gene may be a hotspot for chromosomal breakage. PMID: 11891694
- Congenital disorder of glycosylation type Ia: benign clinical course in a new genetic variant. a new, previously undescribed, combination of mutations of the PMM gene locus on chromosome 16p13 (647,691). PMID: 11935250
- A Japanese patient with congenital disorder of glycosylation type Ia had a novel nonsense mutation (R194X) in the PMM2 gene. PMID: 13129599
- enzymatic activity of PMM2 is upregulated by insulin treatment and that Sgk1 completely inhibits PMM2 activity both in the absence and in the presence of insulin stimulation. PMID: 15342340
- 11 novel mutations in the PMM2 gene are described as associated with Congenital disorder of Glycosylation type Ia. PMID: 15844218
- Based on SNP and STR genotypic analysis, we ascertained an association between the R141H substitution and a particular haplotype, suggesting a common origin for all the mutated chromosomes. PMID: 17166182
- We describe the characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. PMID: 17307006
- three siblings of congenital disorder of glycosylation type 1a are compound heterozygotes for R141H and L32R mutations in the PMM2 gene. PMID: 17451957
- Two CDG-Ia patients,who presented with ataxia and cerebellar hypoplasia and with a normal or only slightly abnormal transferrin but the activity of the corresponding enzyme phosphomannomutase was clearly deficient in leucocytes and fibroblasts. PMID: 17694350
- Val231Met and Arg148Met heterozygosity can present as primary skeletal dysplasia with congenital glycosylation type Ia disorder. PMID: 18203160
- This work reports the study of two new nucleotide changes (c.256-1G>C and c.640-9T>G) identified in the PMM2 gene in congenital disorders of glycosylation type 1a patients, and of a previously described deep intronic nucleotide change in intron 7. PMID: 19235233
顯示更多
收起更多
-
相關(guān)疾病:Congenital disorder of glycosylation 1A (CDG1A)
-
亞細(xì)胞定位:Cytoplasm.
-
蛋白家族:Eukaryotic PMM family
-
數(shù)據(jù)庫(kù)鏈接:
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
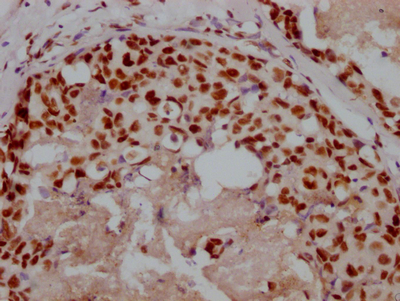
-
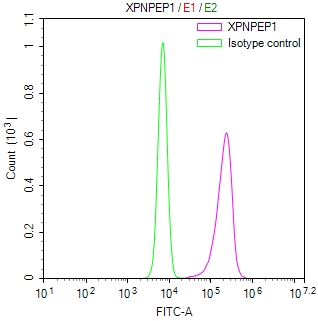
-
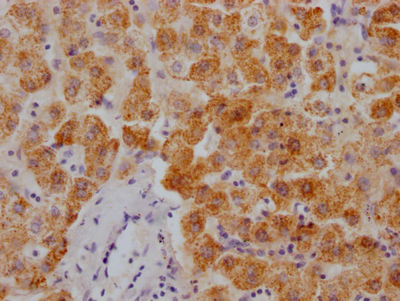
-
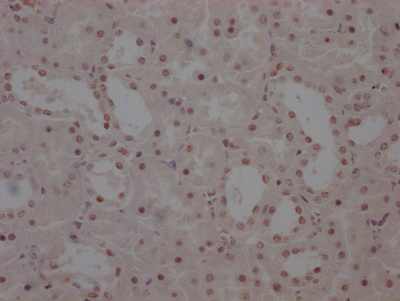
-