FKRP Antibody
-
中文名稱:FKRP兔多克隆抗體
-
貨號:CSB-PA035204
-
規(guī)格:¥2024
-
圖片:
-
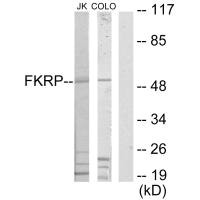 Western blot analysis of extracts from Jurkat cells and COLO205 cells, using FKRP antibody.
Western blot analysis of extracts from Jurkat cells and COLO205 cells, using FKRP antibody.
-
-
其他:
產(chǎn)品詳情
-
產(chǎn)品名稱:Rabbit anti-Homo sapiens (Human) FKRP Polyclonal antibody
-
Uniprot No.:
-
基因名:
-
宿主:Rabbit
-
反應(yīng)種屬:Human,Mouse,Rat
-
免疫原:Synthesized peptide derived from N-terminal of Human FKRP.
-
免疫原種屬:Homo sapiens (Human)
-
克隆類型:Polyclonal
-
純化方式:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
-
濃度:It differs from different batches. Please contact us to confirm it.
-
產(chǎn)品提供形式:Liquid
-
應(yīng)用范圍:ELISA,WB
-
推薦稀釋比:
Application Recommended Dilution WB 1:500-1:3000 -
Protocols:
-
儲存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產(chǎn)品評價

相關(guān)產(chǎn)品
靶點詳情
-
功能:Catalyzes the transfer of CDP-ribitol to ribitol 5-phosphate previously attached by FKTN/fukutin of to the phosphorylated O-mannosyl trisaccharide (N-acetylgalactosamine-beta-3-N-acetylglucosamine-beta-4-(phosphate-6-)mannose), a carbohydrate structure present in alpha-dystroglycan (DAG1). This constitutes the second step in the formation of the ribose 5-phosphate tandem repeat which links the phosphorylated O-mannosyl trisaccharide to the ligand binding moiety composed of repeats of 3-xylosyl-alpha-1,3-glucuronic acid-beta-1.
-
基因功能參考文獻:
- The FKRP-related disorders should be included as a differential diagnosis in Mexican patients with neuromuscular disorders and normal results on DMD gene deletion/duplication analysis. This and previous reports, along with data from the main genotype databases, collectively suggest that in Mexico the FKRP-related disorders are due to the p.(Asn463Asp) and the common European p.(Leu276Ile) pathogenic variants. PMID: 29065428
- The results suggest that fukutin and FKRP not only participate in the synthesis of O-mannosyl glycans added to alpha-dystroglycan in the endoplasmic reticulum and Golgi complex, but that they could also play a role, that remains to be established, in the nucleus of retinal neurons. PMID: 29416295
- This literature review revealed that pathogenic mutations in the FKRP gene in Asian LGMD2I patients are compound heterozygous rather than homozygous. PMID: 28931339
- Next generation and Sanger sequencing were performed for I-2. Heterozygous FKRP mutations were identified in exon 4: c.1167_1168delGC, p.Gly391Leufs *72 and c.501_502GT>CC, p.Arg167Ser, p.Cys168Arg PMID: 28629604
- Fukutin, FKRP, and TMEM5 form a complex while maintaining each of their enzyme activities. Data showed that endogenous fukutin and FKRP enzyme activities coexist with TMEM5 enzyme activity, and suggest the possibility that formation of this enzyme complex may contribute to specific and prompt biosynthesis of glycans that are required for dystroglycan function. PMID: 29477842
- This study have demonstrated that the clinical heterogeneity in LGMD2I patients, homozygous for FKRP c.826C>A, cannot be explained by histopathological alterations, levels of alpha-DG hypoglycosylation or laminin alpha2 depletion. PMID: 28479227
- Dystrophic Pathology in Diaphragm and Impairment of Cardiac Function in FKRP P448L Mutant Mice PMID: 27711214
- The 13 novel mutations of FKRP significantly expanded the mutation spectrum of MDC1C and LGMD2I, and the different founder mutations indicate the ethnic difference in FKRP mutations. PMID: 27439679
- Fukutin and fukutin-related protein are sequentially acting Rbo5P transferases that use cytidine diphosphate ribitol. PMID: 26923585
- Muscular dystrophies can present with rhabdomyolysis; FKRP mutations are particularly frequent in causing such complication. PMID: 26810512
- This provide a new mouse model of Limb-Girdle Muscular Dystrophy Type 2I homozygous for the Common L276I Mutation. PMID: 26574668
- This study demonistrated that the higher frequency of LGMD2I with cardiomyopathy in mutation of FKRP in Taiwanese patients. PMID: 23800702
- FKRP co-localises with the middle-to-trans-Golgi marker MG160, between the myofibrils in human rectus femoris muscle fibres. PMID: 21886772
- Mutations in FKRP lead to a glycosylation defect and subsequently downregulation of alpha-dystroglycan which constitutes an essential component of the proteoglycan-dystrophin complex. PMID: 21311896
- Study revealed a large homozygous block at the LGMD2I locus, and direct sequencing of FKRP encoding fukutin-related-protein detected the common homozygous c.826 C>A (p.Leu276Ile) mutation. PMID: 21172462
- Two novel heterozygous mutations (c.208T>A and c.1030G>T) in the FKRP gene were identified in Chinese brothers with progressive shoulder and pelvic muscle weakness PMID: 21296577
- This study identified FKRP mutations on both alleles in 88 patients from 69 families with Limb Girdle Muscular Dystrophy Type 2I. PMID: 20961759
- two siblings carrying a homozygous mutation in the start codon of FKRP that is likely to result in a loss of functional FKRP protein. The clinical phenotype of the patients was consistent with Walker-Warburg syndrome PMID: 20236121
- our study confirms that typical clinical symptoms (calf hypertrophy, cardiac involvement, mild LGMD) of LGMD2I due the homozygous c.826C[A mutation, are rather frequent in Germany PMID: 19820980
- Co-injection of fish or human FKRP mRNA along with the morpholino restored normal development and alpha-dystroglycan glycosylation. PMID: 19955119
- Alteration of the secretion pathway by different mutations may contribute to wide variations in phenotypes associated with FKRP-related diseases such as muscular dystrophy. PMID: 19900540
- FKRP mutations can result in muscular dystrophy with mental retardation & cerebellar cysts, adding structural brain defects to the FKRP mutation spectrum. Depletion of alpha-dystroglycan expression suggests FKRP involvement in its processing. PMID: 12654965
- patients with mutations in the fukutin-related protein (FKPR) gene had congenital muscular dystrophy PMID: 12666124
- particular FKRP mutations in the homozygous state induce structural and clinical neurological lesions in addition to muscular dystrophy PMID: 14652796
- Data suggest that fukutin and fukutin-related protein (FKRP) may be involved at different steps in O-mannosylglycan synthesis of alpha-dystroglycan, and FKRP is most likely involved in the initial step in this synthesis. PMID: 15213246
- pathogenesis of congenital muscular dystrophies, severity is related to ability to transport protein to ER PMID: 15574464
- a type of LGMD in the Hutterite population maps to chromosome 19q31-q33 and is due to homozygosity for the L276I mutation in FKRP. PMID: 15580560
- Three siblings without clinical signs of muscle dystrophy, but with dilated cardiomyopathy have C826A Fukutin-related protein mutation. PMID: 15833432
- A FKRP point mutation, L276I has been found in all patients with LGMD2I studied so far. The authors screened for this mutation in 102 sporadic cases of Duchenne/Becker mutation-negative patients and found 13 patients with LGMD2I. PMID: 15883334
- Retention in the endoplasmic reticulum of FKRP is not the main mechanism of disease; this may instead relate to a disruption of the functional activity of this putative enzyme with its substrate(s) in the Golgi. PMID: 16055117
- FKRP mutations are a frequent cause of limb-girdle muscular dystrophies. The degree of respiratory and cardiac insufficiency in patients did not correlate with the severity of muscle involvement. PMID: 16344347
- A novel FKRP gene((c.823C>T (p.R275C) and c.948delC)mutation in a Taiwanese patient with limb-girdle muscular dystrophy 2I PMID: 17055682
- We report a limb-girdle muscular dystrophy 2I family with three affected sisters and a highly variable clinical course. FKRP gene sequencing showed that all three sisters carried a nonsense paternal mutation (W225X). PMID: 17113772
- two unrelated Mexican children with congenital muscular dystrophy who each have the identical, novel 1387A>G, N463D mutation. PMID: 17336067
- 3 new FKRP mutations were identified: L322V, L489R and R275G. PMID: 17351538
- Limb-girdle muscular dystrophy (LGMD) type 2I, caused by mutations in the fukutin-related protein gene (FKRP). PMID: 17446099
- The unfolded protein response is activated in LGMD2I muscle biopsies in limb girdle muscular dystrophy type 2I. PMID: 17952692
- severe dilated cardiomyopathy requiring heart transplantation in a homozygous p.Leu276Ile mutation in Fukutin-related protein gene (FKRP) PMID: 18060779
- findings detected a homozygous mutation of the FKRP gene (826C>A) in two unrelated patients with limb-girdle muscular dystrophy 2I with necrotic myopathy with numerous rimmed vacuoles PMID: 18593008
- In our population of LGMD2I patients, different mutations in the FKRP gene are associated with several secondary muscle protein reductions, and the deficiencies of alpha2-laminin and alpha-DG on sections are prevalent. PMID: 18645206
- Data show that four sibs belonging to a second Tunisian LGMD2I family show variable cardiac involvement with FKRP gene mutations. PMID: 18671187
- mutation spectrum associted with limb-girdle muscular dystrophy variability and serverity PMID: 19917824
- Limb girdle muscular dystrophy 2I is a milder allelic variant of MDC1C. PMID: 11741828
顯示更多
收起更多
-
相關(guān)疾病:Muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A5 (MDDGA5); Muscular dystrophy-dystroglycanopathy congenital with or without mental retardation B5 (MDDGB5); Muscular dystrophy-dystroglycanopathy limb-girdle C5 (MDDGC5)
-
亞細胞定位:Golgi apparatus membrane; Single-pass type II membrane protein. Secreted. Cell membrane, sarcolemma. Rough endoplasmic reticulum. Cytoplasm.
-
蛋白家族:LicD transferase family
-
組織特異性:Expressed in the retina (at protein level). Expressed predominantly in skeletal muscle, placenta, and heart and relatively weakly in brain, lung, liver, kidney, and pancreas.
-
數(shù)據(jù)庫鏈接:
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-