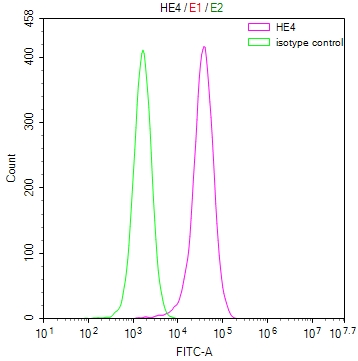
ALS2 Antibody
-
中文名稱:ALS2兔多克隆抗體
-
貨號:CSB-PA857009ESR2HU
-
規格:¥440
-
圖片:
-
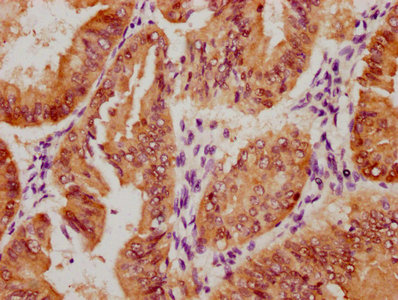
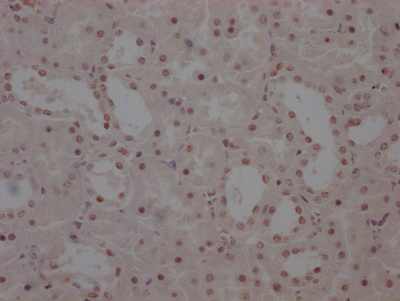
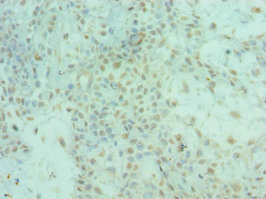 Immunohistochemistry of paraffin-embedded human breast cancer using CSB-PA857009ESR2HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human breast cancer using CSB-PA857009ESR2HU at dilution of 1:100 -
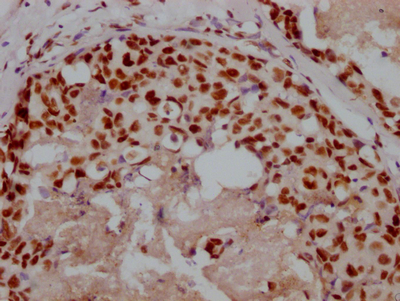
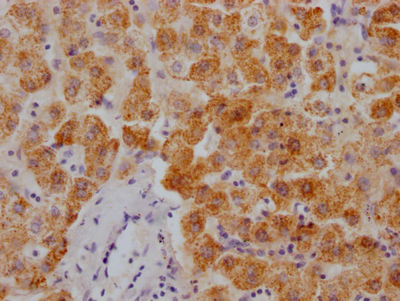
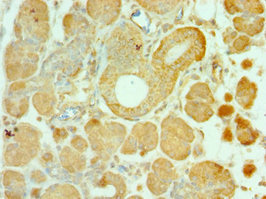 Immunohistochemistry of paraffin-embedded human pancreatic tissue using CSB-PA857009ESR2HU at dilution of 1:100
Immunohistochemistry of paraffin-embedded human pancreatic tissue using CSB-PA857009ESR2HU at dilution of 1:100
-
-
其他:
產品詳情
-
產品名稱:Rabbit anti-Homo sapiens (Human) ALS2 Polyclonal antibody
-
Uniprot No.:
-
基因名:ALS2
-
別名:ALS 2 antibody; ALS2 antibody; ALS2_HUMAN antibody; ALS2CR6 antibody; Alsin antibody; ALSJ antibody; Amyotrophic lateral sclerosis 2 (juvenile) antibody; Amyotrophic lateral sclerosis 2 (juvenile) chromosome region candidate 6 antibody; Amyotrophic lateral sclerosis 2 chromosomal region candidate gene 6 protein antibody; Amyotrophic lateral sclerosis 2 protein antibody; Amyotrophic lateral sclerosis protein 2 antibody; FLJ31851 antibody; IAHSP antibody; KIAA1563 antibody; MGC87187 antibody; PLSJ antibody
-
宿主:Rabbit
-
反應種屬:Human
-
免疫原:Recombinant Human Alsin protein (1-280AA)
-
免疫原種屬:Homo sapiens (Human)
-
標記方式:Non-conjugated
-
克隆類型:Polyclonal
-
抗體亞型:IgG
-
純化方式:Antigen Affinity Purified
-
濃度:It differs from different batches. Please contact us to confirm it.
-
保存緩沖液:PBS with 0.02% sodium azide, 50% glycerol, pH7.3.
-
產品提供形式:Liquid
-
應用范圍:ELISA, IHC
-
推薦稀釋比:
Application Recommended Dilution IHC 1:20-1:200 -
Protocols:
-
儲存條件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
貨期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
-
用途:For Research Use Only. Not for use in diagnostic or therapeutic procedures.
產品評價

相關產品
靶點詳情
-
功能:May act as a GTPase regulator. Controls survival and growth of spinal motoneurons.
-
基因功能參考文獻:
- This study identified a novel ALS2 pathogenic founder variant in Iran that further adds to the allelic heterogeneity of infantile-onset ascending hereditary spastic paralysis. PMID: 30128655
- Nonsense mutation in ALS2 gene is associated with severe and progressive infantile onset of spastic paralysis. PMID: 28502191
- We established a genetic diagnosis in six families with autosomal recessive HSP (SPG11 in three families and TFG/SPG57, SACS and ALS2 in one family each). A heterozygous mutation in a gene involved in an autosomal dominant HSP (ATL1/SPG3A) was also identified in one additional family PMID: 27601211
- This study identified two novel ALS2 mutations in two Pakistani families with infantile-onset ascending hereditary spastic paraplegia cosegregating with the disease. PMID: 26751646
- novel compound heterozygous ALS2 deletion mutations were identified in two siblings with infantile ascending hereditary spastic paraplegia. PMID: 25433428
- We identified a novel homozygous splice-site mutation (c.3512+1G>A) in the ALS2 gene (NM_020919.3) encoding alsin that segregated with the disease in this family PMID: 25474699
- Data indicate a splice-site mutation of the amyotrophic lateral sclerosis 2 (juvenile) protein (ALS2) in four children of a consanguineous family with infantile-onset ascending hereditary spastic paraplegia. PMID: 24704789
- The ALS2 gene should be screened for mutations in patients who present with generalized dystonia and cerebellar signs. PMID: 24562058
- The ALS2 mutation c.2761C>T leading to infantile-onset hereditary spastic paraplegia resides in the pleckstrin domain, which is involved in the overall neuronal development or maintenance. PMID: 24315819
- ALS2 sequencing revealed two heterozygous mutations: the missense variant c.299 G>T, leading to the replacement of a serine with an isoleucine (p.S100I), and the splicing variant c.2580-2 A>G in brothers with juvenile amyotrophic lateral sclerosis. PMID: 23282280
- these results suggest that Als2 is a binding partner of Uxt and Als2/Uxt interaction could be important for the activation of Nf-kappaB pathway. PMID: 21907703
- causative genes for familial amyotrophic lateral sclerosis PMID: 12138710
- Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. PMID: 12145748
- 16 patients from 11 unrelated families were studied with a phenotype of infantile ascending hereditary spastic paralysis (IAHSP); Alsin mutations were found in 4 of the 10 families, whereas haplotype analysis excluded the ALS2 locus in one family PMID: 12601111
- Perturbation of endosomal dynamics caused by loss of ALS2 rab5GEF activity might underlie neuronal dysfunction and degeneration. PMID: 12837691
- deletion mutations in ALS2 gene detected in ALS2 patients seem to be uncommon in Japanese AR-ALS, and that SNPs in uncoding regions might possibly be relevant to predisposition to ALS. PMID: 12866199
- A nonsense mutation in alsin was found in infantile spastic paraplegia. Full-length alsin is probably required for the proper development and/or functioning of upper motor neurons. PMID: 12919135
- Mutations in the ALS2 gene linked to early-onset motor neuron disease uniformly produce loss of activity through decreased protein stability of this endosomal protein. PMID: 14668431
- Mutations of ALS2 are not a common cause of ALS. PMID: 14676054
- Expression of alsin LF, but not alsin short form, protected motor neuronal cells from toxicity induced by mutants of the Cu/Zn-superoxide dismutase (SOD1) gene, which cause autosomal dominant ALS PMID: 14970233
- oligomerization of the ALS2 protein is one of the fundamental features for its physiological function involving endosome dynamics in vivo PMID: 15247254
- A peptide derived from the ALS2 protein is selectively localized to the somatodendritic compartment of motor neurons in human spinal cord. PMID: 15371724
- These results suggest that amyotrophic lateral sclerosis 2 C-terminal like (ALS2CL), a novel ALS2 homologue, modulates Rab5-mediated endosome dynamics in HeLa cells. PMID: 15388334
- Rac1, PI3 kinase, and Akt3 have roles in an anti-apoptotic pathway triggered by ALS2 that antagonizes SOD1 mutant-induced motoneuronal cell death PMID: 15579468
- ALS2/Alsin has a role in regulating Rac-PAK signaling and neurite outgrowth PMID: 16049005
- colocalization of Alsin with the centrosomal markers gamma-tubulin and A kinase anchoring protein. PMID: 16085057
- ALS2 mutations are not implicated in the pathogenesis of adult-onset primary lateral sclerosis. PMID: 17698795
- Autosomal recessive mutations in the ALS2 gene lead to a clinical spectrum of motor dysfunction including juvenile onset amyotrophic lateral sclerosis. [REVIEW] PMID: 17955197
- mutations in ALS2 also need to be considered in patients from northwestern Europe with early-onset spastic paralysis and amyotrophic or primary lateral sclerosis. PMID: 18523452
- Results suggest at least four recombination events in the ALS2 gene during maternal meiosis followed by a meiosis I error and postzygotic trisomy rescue or gamete complementation, in a patient with infantile-onset ascending spastic paralysis. PMID: 18810511
- A structural model for the N-terminal 690-residue region of alsin through comparative modelling based on regulator of chromosome condensation 1 was created. PMID: 19023603
- This novel ALS2 splice-site mutation is causing the loss of exon 18 in the transcript which results in a frameshift after exon 17. PMID: 19122027
顯示更多
收起更多
-
相關疾病:Amyotrophic lateral sclerosis 2 (ALS2); Juvenile primary lateral sclerosis (JPLS); Infantile-onset ascending spastic paralysis (IAHSP)
-
數據庫鏈接:
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-